

这篇文章很长,超过1万字,是本系列中最重要的一篇,因为我并非只是在简单地告诉大家几条硬邦邦的操作命令。对于新手而言不建议碎片时间阅读,对于有一定经验的老手来说,相信依然可以有所收获。在开始之前,我想先说一句:流程的具体形式其实是次要的,WGS本质上只是一个技术手段,重要的是,我们要明白自己所要解决的问题是什么,所希望获取的结果是什么,然后再选择合适的技术。这是许多人经常忽视的一个重要问题。
好了,以下进入正文。
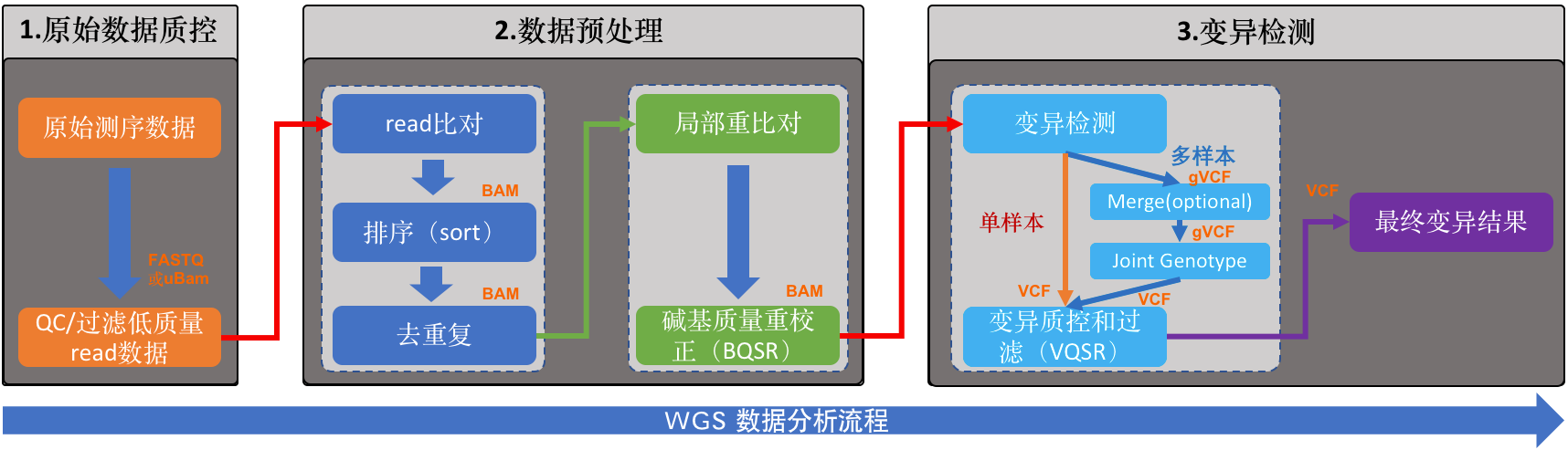
这是WGS数据分析的流程图。流程的目的是准确检测出每个样本(这里特指人)基因组中的变异集合,也就是人与人之间存在差异的那些DNA序列。我把整个分析过程按照它们实际要完成的功能,将其分成了三个大的模块:
- 原始数据质控
- 数据预处理
- 变异检测
这或许和很多人看到的WGS分析流程,在结构梳理上有些差异(比如GATK的最佳实践),但过程中的各个步骤和所要完成的事情是一模一样的。
0.准备阶段
在开始之前,我们需要做一些准备工作,主要是部署好相关的软件和工具。我们在这个WGS数据分析过程中用到的所有软件都是开源的,它们的代码全部都能够在github上找到,具体如下:
- BWA(Burrow-Wheeler Aligner): 这是最权威,使用最广的NGS数据比对软件,目前已经更新到0.7.16版本;
- Samtools: 是一个专门用于处理比对数据的工具,由BWA的作者(lh3)所编写;
- Picard: 它是目前最著名的组学研究中心-Broad研究所开发的一款强大的NGS数据处理工具,功能方面和Samtools有些重叠,但更多的是互补,它是由java编写的,我们直接下载最新的.jar包就行了。
- GATK: 同样是Broad研究所开发的,是目前业内最权威、使用最广的基因数据变异检测工具。值得注意的是,目前GATK有3.x和4.x两个不同的版本,代码在github上也是分开的。4.x是今年新推出的,在核心算法层面并没太多的修改,但使用了新的设计模式,做了很多功能的整合,是更适合于大规模集群和云运算的版本,后续GATK团队也将主要维护4.x的版本,而且它的代码是100%开源的,这和3.x只有部分开源的情况不同。看得出GATK今年的这次升级是为了应对接下来越来越多的大规模人群测序数据而做出的改变,但现阶段4.x版本还不稳定,真正在使用的人和机构其实也还不多。短期来看,3.x版本还将在业内继续使用一段时间;其次,3.x对于绝大分部的分析需求来说是完全足够的。我们在这里也以GATK3.8(最新版本)作为流程的重要工具进行分析流程的构建。
事实上,对于构造WGS分析流程来说,以上这个四个工具就完全足够了。它们的安装都非常简单,除了BWA和Samtools由C编写的,安装时需要进行编译之外,另外两个只要保证系统中的java是1.8.x版本及以上的,那么直接下载jar包就可以使用了。操作系统方面推荐linux(集群)或者Mac OS。
1.原始数据质控
数据的质控,由于我已经在上一节的文章中讲的比较详细了,因此在本篇中就不再进行详细的讨论了。而且质控的处理方法都是比较一致的,基本不需要为特定的分析做定制化的改动,因此,我们可以把它作为WGS主流程之外的一环。但还是再强调一下,数据质控的地位同样重要,不然我也不必专门为其单独写一篇完整的文章。
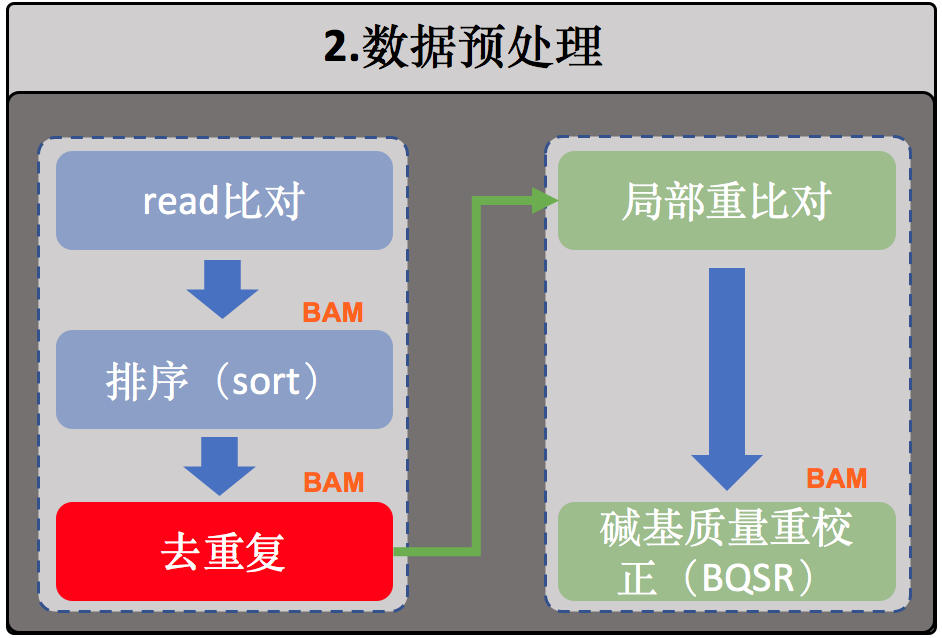
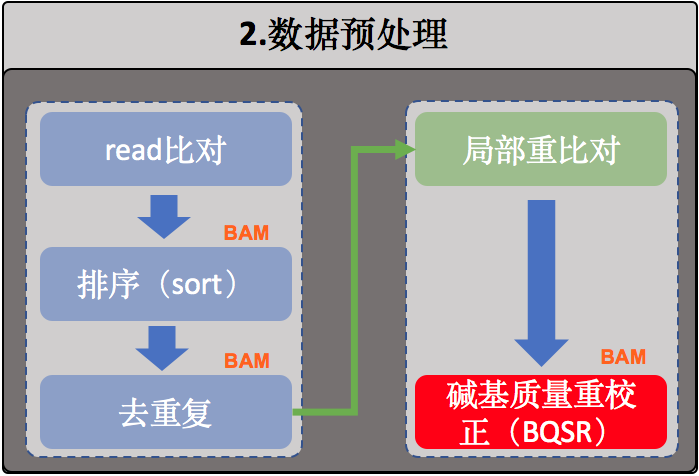
2.数据预处理
序列比对
先问一个问题:为什么需要比对?
我们已经知道NGS测序下来的短序列(read)存储于FASTQ文件里面。虽然它们原本都来自于有序的基因组,但在经过DNA建库和测序之后,文件中不同read之间的前后顺序关系就已经全部丢失了。因此,FASTQ文件中紧挨着的两条read之间没有任何位置关系,它们都是随机来自于原本基因组中某个位置的短序列而已。
因此,我们需要先把这一大堆的短序列捋顺,一个个去跟该物种的 参考基因组【注】比较,找到每一条read在参考基因组上的位置,然后按顺序排列好,这个过程就称为测序数据的比对。这 也是核心流程真正意义上的第一步,只有完成了这个序列比对我们才有下一步的数据分析。
【注】参考基因组:指该物种的基因组序列,是已经组装成的完整基因组序列,常作为该物种的标准参照物,比如人类基因组参考序列,fasta格式。
序列比对本质上是一个寻找最大公共子字符串的过程。大家如果有学过生物信息学的话,应该或多或少知道BLAST,它使用的是动态规划的算法来寻找这样的子串,但在面对巨量的短序列数据时,类似BLAST这样的软件实在太慢了!因此,需要更加有效的数据结构和相应的算法来完成这个搜索定位的任务。
我们这里将用于流程构建的BWA就是其中最优秀的一个,它将BW(Burrows-Wheeler)压缩算法和后缀树相结合,能够让我们以较小的时间和空间代价,获得准确的序列比对结果。
以下我们就开始流程的搭建。
首先,我们需要为参考基因组的构建索引——这其实是在为参考序列进行Burrows Wheeler变换(wiki: 块排序压缩),以便能够在序列比对的时候进行快速的搜索和定位。
- $ bwa index human.fasta
以我们人类的参考基因组(3Gb长度)为例,这个构造过程需要消耗几个小时的时间(一般3个小时左右)。完成之后,你会看到类似如下几个以human.fasta为前缀的文件:
- .
- ├── human.fasta.amb
- ├── human.fasta.ann
- ├── human.fasta.bwt
- ├── human.fasta.pac
- └── human.fasta.sa
这些就是在比对时真正需要被用到的文件。这一步完成之后,我们就可以将read比对至参考基因组了:
- $ bwa mem -t 4 -R '@RG\tID:foo_lane\tPL:illumina\tLB:library\tSM:sample_name' /path/to/human.fasta read_1.fq.gz read_2.fq.gz > sample_name.sam
大伙如果以前没使用过这个比对工具的话,那么可能不明白上面参数的含义。我们这里调用的是bwa的mem比对模块,在解释这样做之前,我们不妨先看一下bwa mem的官方用法说明,它就一句话:
- Usage: bwa mem [options] <idxbase> <in1.fq> [in2.fq]
其中,[options]是一系列可选的参数,暂时不多说。这里的 < idxbase>要输入的是参考基因组的BW索引文件,我们上面通过bwa index构建好的那几个以human.fasta为前缀的文件便是;< in1.fq>和 [in2.fq]输入的是质控后的fastq文件。但这里输入的时候为什么会需要两个fq(in1.fq和in2.fq)呢?我们上面的例子也是有两个:read_1.fq.gz和read_2.fq.gz。这是因为这是双末端测序(也称Pair-End)的情况,那什么是“双末端测序”呢?这两个fq之间的关系又是什么?这个我需要简单解析一下。
我们已经知道NGS是短读长的测序技术,一次测出来的read的长度都不会太长,那为了尽可能把一个DNA序列片段尽可能多地测出来,既然测一边不够,那就测两边,于是就有了一种从被测DNA序列两端各测序一次的模式,这就被称为双末端测序(Pair-End Sequencing,简称PE测序)。如下图是Pair-End测序的示意图,中间灰色的是被测序的DNA序列片段,左边黄色带箭头和右边蓝色带箭头的分别是测序出来的read1和read2序列,这里假定它们的长度都是100bp。虽然很多时候Pair-End测序还是无法将整个被测的DNA片段完全测通,但是它依然提供了极其有用的信息,比如,我们知道每一对的read1和read2都来自于同一个DNA片段,read1和read2之间的距离是这个DNA片段的长度,而且read1和read2的方向刚好是相反的(这里排除mate-pair的情形)等,这些信息对于后面的变异检测等分析来说都是非常有用的。
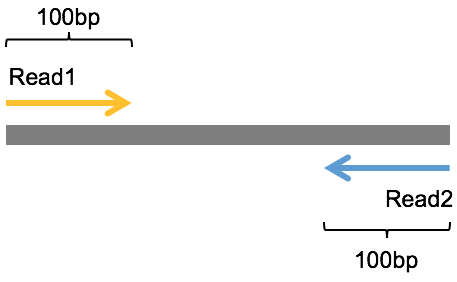
另外,在read1在fq1文件中位置和read2在fq2文件中的文件中的位置是相同的,而且read ID之间只在末尾有一个’/1’或者’/2’的差别。
既然有双末端测序,那么与之对应的就有单末端测序(Single End Sequecing,简称SE测序),即只测序其中一端。因此,我们在使用bwa比对的时候,实际上,in2.fq是非强制性的(所以用方括号括起来),只有是双末端测序的数据时才需要添加。
回到上面我们的例子,大伙可以看到我这里除了用法中提到的参数之外,还多了2个额外的参数,分别是:-t,线程数,我们在这里使用4个线程;-R 接的是 Read Group的字符串信息,这是一个非常重要的信息,以@RG开头,它是用来将比对的read进行分组的。不同的组之间测序过程被认为是相互独立的,这个信息对于我们后续对比对数据进行错误率分析和Mark duplicate时非常重要。在Read Group中,有如下几个信息非常重要:
(1) ID,这是Read Group的分组ID,一般设置为测序的lane ID(不同lane之间的测序过程认为是独立的),下机数据中我们都能看到这个信息的,一般都是包含在fastq的文件名中;
(2) PL,指的是所用的测序平台,这个信息不要随便写!特别是当我们需要使用GATK进行后续分析的时候,更是如此!这是一个很多新手都容易忽视的一个地方,在GATK中,PL只允许被设置为:ILLUMINA,SLX,SOLEXA,SOLID,454,LS454,COMPLETE,PACBIO,IONTORRENT,CAPILLARY,HELICOS或UNKNOWN这几个信息。基本上就是目前市场上存在着的测序平台,当然,如果实在不知道,那么必须设置为UNKNOWN,名字方面不区分大小写。如果你在分析的时候这里没设置正确,那么在后续使用GATK过程中可能会碰到类似如下的错误:
- ERROR MESSAGE: The platform (xx) associated with read group GATKSAMReadGroupRecord @RG:xx is not a recognized platform.
这个时候你需要对比对文件的header信息进行重写,就会稍微比较麻烦。
我们上面的例子用的是PL:illumina。如果你的数据是CG测序的那么记得不要写成CG!而要写COMPLETE。
(3) SM,样本ID,同样非常重要,有时候我们测序的数据比较多的时候,那么可能会分成多个不同的lane分布测出来,这个时候SM名字就是可以用于区分这些样本。
(4) LB,测序文库的名字,这个重要性稍微低一些,主要也是为了协助区分不同的group而存在。文库名字一般可以在下机的fq文件名中找到,如果上面的lane ID足够用于区分的话,也可以不用设置LB;
除了以上这四个之外,还可以自定义添加其他的信息,不过如无特殊的需要,对于序列比对而言,这4个就足够了。这些信息设置好之后,在RG字符串中要用制表符(\t)将它们分开。
最后在我们的例子中,我们将比对的输出结果直接重定向到一份sample_name.sam文件中,这类文件是BWA比对的标准输出文件,它的具体格式我会在下一篇文章中进行详细说明。但SAM文件是文本文件,一般整个文件都非常巨大,因此,为了有效节省磁盘空间,一般都会用samtools将它转化为BAM文件(SAM的特殊二进制格式),而且BAM会更加方便于后续的分析。所以我们上面比对的命令可以和samtools结合并改进为:
- $ bwa mem -t 4 -R '@RG\tID:foo_lane\tPL:illumina\tLB:library\tSM:sample_name' /path/to/human.fasta read_1.fq.gz read_2.fq.gz | samtools view -S -b - > sample_name.bam
我们通过管道(“|”)把比对的输出如同引导水流一样导流给samtools去处理,上面samtools view的-b参数指的就是输出为BAM文件,这里需要注意的地方是-b后面的’-‘,它代表就是上面管道引流过来的数据,经过samtools转换之后我们再重定向为sample_name.bam。
关于BWA的其他参数,我这里不打算对其进行一一解释,在绝大多数情况下,采用默认是合适的做法。
[Tips] BWA MEM比对模块是有一定适用范围的:它是专门为长read比对设计的,目的是为了解决,第三代测序技术这种能够产生长达几十kb甚至几Mbp的read情况。一般只有当read长度≥70bp的时候,才推荐使用,如果比这个要小,建议使用BWA ALN模块。
排序
以上,我们就完成了read比对的步骤。接下来是排序:
排序这一步我们也是通过使用samtools来完成的,命令很简单:
- Usage: samtools sort [options...] [in.bam]
但在执行之前,我们有必要先搞明白为什么需要排序,为什么BWA比对后输出的BAM文件是没顺序的!原因就是FASTQ文件里面这些被测序下来的read是随机分布于基因组上面的,第一步的比对是按照FASTQ文件的顺序把read逐一定位到参考基因组上之后,随即就输出了,它不会也不可能在这一步里面能够自动识别比对位置的先后位置重排比对结果。因此,比对后得到的结果文件中,每一条记录之间位置的先后顺序是乱的,我们后续去重复等步骤都需要在比对记录按照顺序从小到大排序下来才能进行,所以这才是需要进行排序的原因。对于我们的例子来说,这个排序的命令如下:
- $ time samtools sort -@ 4 -m 4G -O bam -o sample_name.sorted.bam sample_name.bam
其中,-@,用于设定排序时的线程数,我们设为4;-m,限制排序时最大的内存消耗,这里设为4GB;-O 指定输出为bam格式;-o 是输出文件的名字,这里叫sample_name.sorted.bam。我会比较建议大伙在做类似分析的时候在文件名字将所做的关键操作包含进去,因为这样即使过了很长时间,当你再去看这个文件的时候也能够立刻知道当时对它做了什么;最后就是输入文件——sample_name.bam。
【注意】排序后如果发现新的BAM文件比原来的BAM文件稍微小一些,不用觉得惊讶,这是压缩算法导致的结果,文件内容是没有损失的。
去除重复序列(或者标记重复序列)
在排序完成之后我们就可以开始执行去除重复(准确来说是 去除PCR重复序列)的步骤了。
首先,我们需要先理解什么是重复序列,它是如何产生的,以及为什么需要去除掉?要回答这几个问题,我们需要再次理解在建库和测序时到底发生了什么。
我们在第1节中已经知道,在NGS测序之前都需要先构建测序文库:通过物理(超声)打断或者化学试剂(酶切)切断原始的DNA序列,然后选择特定长度范围的序列去进行PCR扩增并上机测序。
因此,这里重复序列的来源实际上就是由PCR过程中所引入的。因为所谓的PCR扩增就是把原来的一段DNA序列复制多次。可是为什么需要PCR扩增呢?如果没有扩增不就可以省略这一步了吗?
情况确实如此,但是很多时候我们构建测序文库时能用的细胞量并不会非常充足,而且在打断的步骤中也会引起部分DNA的降解,这两点会使整体或者局部的DNA浓度过低,这时如果直接从这个溶液中取样去测序就很可能漏掉原本基因组上的一些DNA片段,导致测序不全。而PCR扩增的作用就是为了把这些微弱的DNA多复制几倍乃至几十倍,以便增大它们在溶液中分布的密度,使得能够在取样时被获取到。所以这里大家需要记住一个重点,PCR扩增原本的目的是为了增大微弱DNA序列片段的密度,但由于整个反应都在一个试管中进行,因此其他一些密度并不低的DNA片段也会被同步放大,那么这时在取样去上机测序的时候,这些DNA片段就很可能会被重复取到相同的几条去进行测序(下图为PCR扩增示意图)。
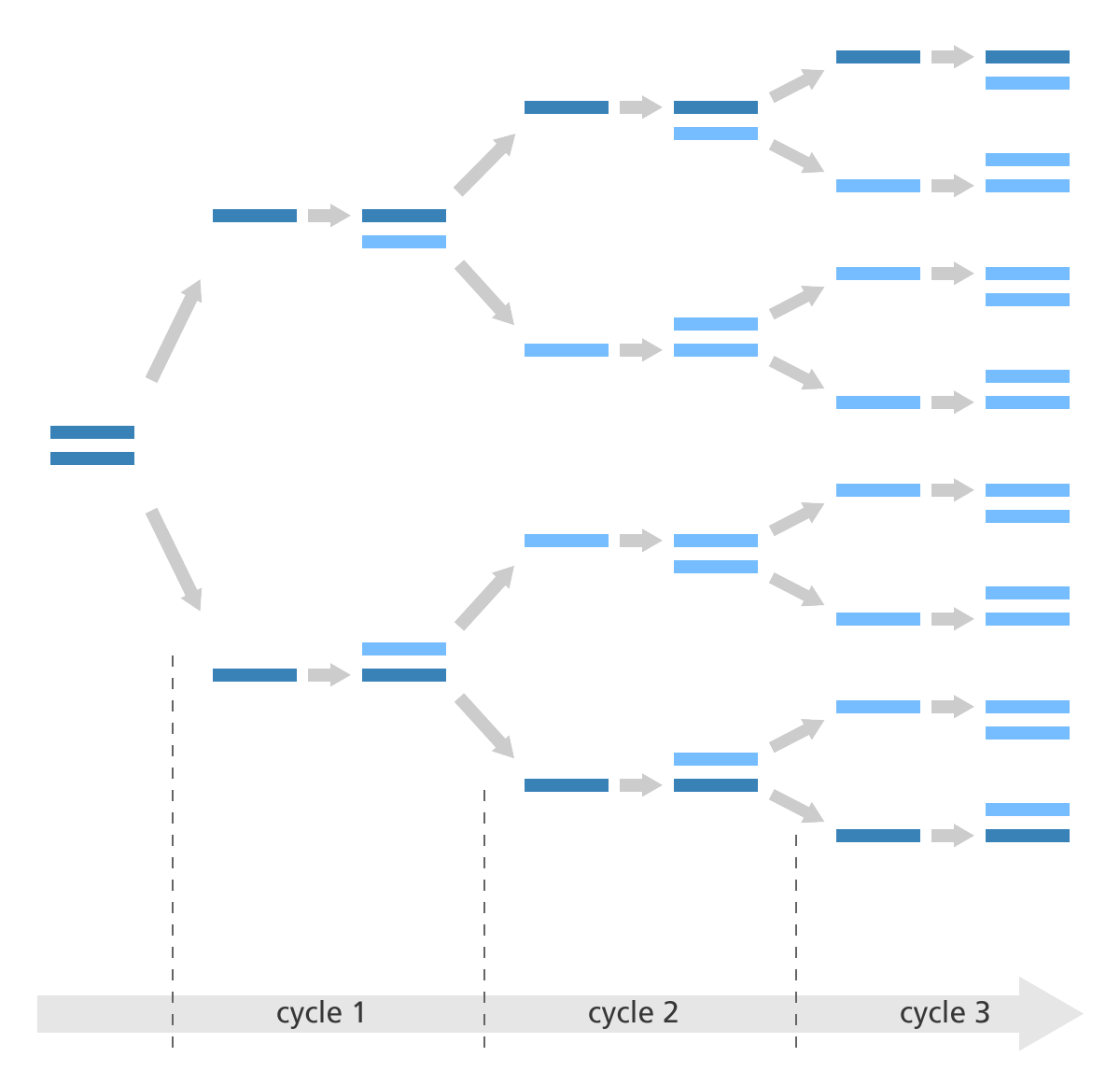
PCR扩增示意图:PCR扩增是一个指数扩增的过程,图中原本只有一段双链DNA序列,在经过3轮PCR后就被扩增成了8段
看到这里,你或许会觉得,那没必要去除不也应该可以吗?因为即便扩增了多几次,不也同样还是原来的那一段DNA吗?直接用来分析对结果也不会有影响啊!难道不是吗?
会有影响,而且有时影响会很大!最直接的后果就是同时增大了变异检测结果的假阴和假阳率。主要有几个原因:
- DNA在打断的那一步会发生一些损失,主要表现是会引发一些碱基发生颠换变换(嘌呤-变嘧啶或者嘧啶变嘌呤),带来假的变异。PCR过程会扩大这个信号,导致最后的检测结果中混入了假的结果;
- PCR反应过程中也会带来新的碱基错误。发生在前几轮的PCR扩增发生的错误会在后续的PCR过程中扩大,同样带来假的变异;
- 对于真实的变异,PCR反应可能会对包含某一个碱基的DNA模版扩增更加剧烈(这个现象称为PCR Bias)。如果反应体系是对含有reference allele的模板扩增偏向强烈,那么变异碱基的信息会变小,从而会导致假阴。
PCR对真实的变异检测和个体的基因型判断都有不好的影响。GATK、Samtools、Platpus等这种利用贝叶斯原理的变异检测算法都是认为所用的序列数据都不是重复序列(即将它们和其他序列一视同仁地进行变异的判断,所以带来误导),因此必须要进行标记(去除)或者使用PCR-Free的测序方案(这个方案目前正变得越来越流行,特别是对于RNA-Seq来说尤为重要,现在著名的基因组学研究所——Broad Institute,基本都是使用PCR-Free的测序方案)。
那么具体是如何做到去除这些PCR重复序列的呢?我们可以抛开任何工具,仔细想想,既然PCR扩增是把同一段DNA序列复制出很多份,那么这些序列在经过比对之后它们一定会定位到基因组上相同的位置,比对的信息看起来也将是一样的!于是,我们就可以根据这个特点找到这些重复序列了!
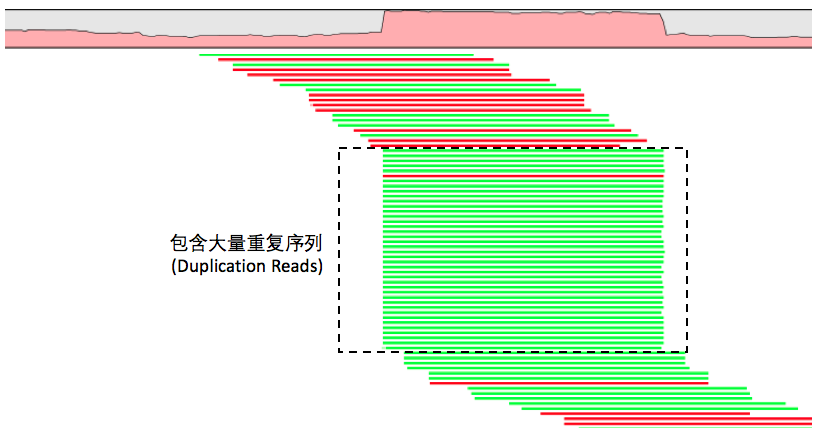
事实上,现有的工具包括Samtools和Picard中去除重复序列的算法也的确是这么做的。不同的地方在于,samtools的rmdup是直接将这些重复序列从比对BAM文件中删除掉,而Picard的MarkDuplicates默认情况则只是在BAM的FLAG信息中标记出来,而不是删除,因此这些重复序列依然会被留在文件中,只是我们可以在变异检测的时候识别到它们,并进行忽略。
考虑到尽可能和现在主流的做法一致(但我并不是说主流的做法就一定是对的,要分情况看待,只是主流的做法容易被做成生产流程而已),我们这里也用Picard来完成这个事情:
- java -jar picard.jar MarkDuplicates \
- I=sample_name.sorted.bam \
- O=sample_name.sorted.markdup.bam \
- M=sample_name.markdup_metrics.txt
这里只把重复序列在输出的新结果中标记出来,但不删除。如果我们非要把这些序列完全删除的话可以这样做:
- java -jar picard.jar MarkDuplicates \
- REMOVE_DUPLICATES=true \
- I=sample_name.sorted.bam \
- O=sample_name.sorted.markdup.bam \
- M=sample_name.markdup_metrics.txt
把参数REMOVE_DUPLICATES设置为ture,那么重复序列就被删除掉,不会在结果文件中留存。我比较建议使用第一种做法,只是标记出来,并留存这些序列,以便在你需要的时候还可以对其做分析。
这一步完成之后,我们需要为sample_name.sorted.markdup.bam创建索引文件,它的作用能够让我们可以随机访问这个文件中的任意位置,而且后面的“局部重比对”步骤也要求这个BAM文件一定要有索引,命令如下:
- $ samtools index sample_name.sorted.markdup.bam
完成之后,会生成一份sample_name.sorted.markdup.bam.bai文件,这就是上面这份BAM的index。
局部重比对
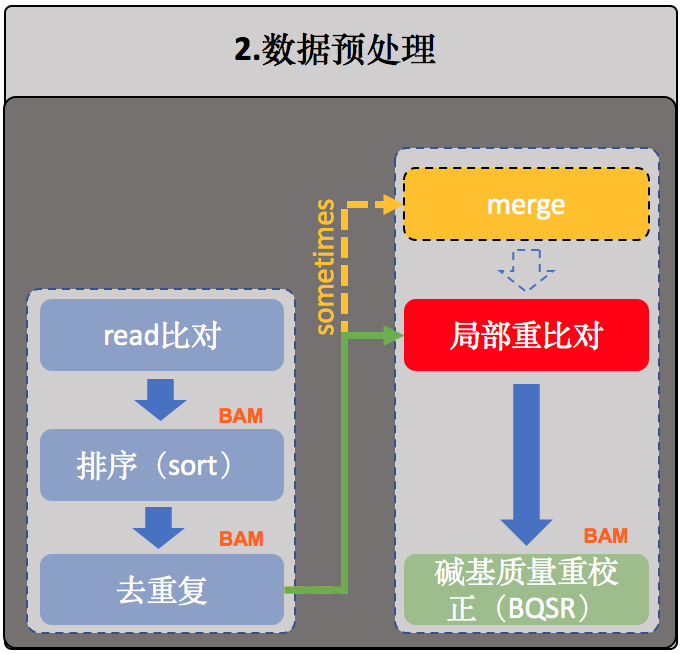
接下来是局部区域重比对,通常也叫Indel局部区域重比对。有时在进行这一步骤之前还有一个merge的操作,将同个样本的所有比对结果合并成唯一一个大的BAM文件【注】,merge的例子如下:
- $ samtools merge <out.bam> <in1.bam> [<in2.bam> ... <inN.bam>]
【注意】之所以会有这种情况,是因为有些样本测得非常深,其测序结果需要经过多次测序(或者分布在多个不同的测序lane中)才全部获得,这个时候我们一般会先分别进行比对并去除重复序列后再使用samtools进行合并。
局部重比对的目的是将BWA比对过程中所发现有 潜在序列插入或者序列删除(insertion和deletion,简称Indel)的区域进行重新校正。这个过程往往还会把一些已知的Indel区域一并作为重比对的区域,但为什么需要进行这个校正呢?
其根本原因来自于参考基因组的序列特点和BWA这类比对算法本身,注意这里不是针对BWA,而是针对所有的这类比对算法,包括bowtie等。这类在全局搜索最优匹配的算法在存在Indel的区域及其附近的比对情况往往不是很准确,特别是当一些存在长Indel、重复性序列的区域或者存在长串单一碱基(比如,一长串的TTTT或者AAAAA等)的区域中更是如此。
另一个重要的原因是在这些比对算法中,对碱基错配和开gap的容忍度是不同的。具体体现在罚分矩阵的偏向上,例如,在read比对时,如果发现碱基错配和开gap都可以的话,它们会更偏向于错配。但是这种偏向错配的方式,有时候却还会反过来引起错误的开gap!这就会导致基因组上原本应该是一个长度比较大的Indel的地方,被错误地切割成多个错配和短indel的混合集,这必然会让我们检测到很多错误的变异。而且,这种情况还会随着所比对的read长度的增长(比如三代测序的Read,通常都有几十kbp)而变得越加严重。
因此,我们需要有一种算法来对这些区域进行局部的序列重比对。这个算法通常就是大名鼎鼎的Smith-Waterman算法,它非常适合于这类场景,可以极其有效地实现对全局比对结果的校正和调整,最大程度低地降低由全局比对算法的不足而带来的错误。而且GATK的局部重比对模块,除了应用这个算法之外,还会对这个区域中的read进行一次局部组装,把它们连接成为长度更大的序列,这样能够更进一步提高局部重比对的准确性。
下图给大家展示一个序列重比对之前和之后的结果,其中灰色的横条指的是read,空白黑线指的是deletion,有颜色的碱基指的是错配碱基。

相信大家都可以明显地看到在序列重比对之前,在这个区域的比对数据是多么的糟糕,如果就这样进行变异检测,那么一定会得到很多假的结果。而在经过局部重比对之后,这个区域就变得非常清晰而分明,它原本发生的就只是一个比较长的序列删除(deletion)事件,但在原始的比对结果中却被错误地用碱基错配和短的Indel所代替。
说到这里,那么具体该怎么做呢?我们的WGS分析流程从这个步骤开始就需要用到GATK (GenomeAnalysisTK.jar)了,我们的执行命令如下:
- java -jar /path/to/GenomeAnalysisTK.jar \
- -T RealignerTargetCreator \
- -R /path/to/human.fasta \
- -I sample_name.sorted.markdup.bam \
- -known /path/to/gatk/bundle/1000G_phase1.indels.b37.vcf \
- -known /path/to/gatk/bundle/Mills_and_1000G_gold_standard.indels.b37.vcf \
- -o sample_name.IndelRealigner.intervals
- java -jar /path/to/GenomeAnalysisTK.jar \
- -T IndelRealigner \
- -R /path/to/human.fasta \
- -I sample_name.sorted.markdup.bam \
- -known /path/to/gatk/bundle/1000G_phase1.indels.b37.vcf \
- -known /path/to/gatk/bundle/Mills_and_1000G_gold_standard.indels.b37.vcf \
- -o sample_name.sorted.markdup.realign.bam \
- --targetIntervals sample_name.IndelRealigner.intervals
这里包含了两个步骤:
- 第一步,RealignerTargetCreator ,目的是定位出所有需要进行序列重比对的目标区域(如下图);
- 第二步,IndelRealigner,对所有在第一步中找到的目标区域运用算法进行序列重比对,最后得到捋顺了的新结果。
IndelRealigner.intervals文件内容示例
以上这两个步骤是缺一不可的,顺序也是固定的。而且,需要指出的是,这里的-R参数输入的human.fasta不是BWA比对中的索引文件前缀,而是参考基因组序列(FASTA格式)文件,下同。
另外,在重比对步骤中,我们还看到了两个陌生的VCF文件,分别是:1000G_phase1.indels.b37.vcf和Mills_and_1000G_gold_standard.indels.b37.vcf。这两个文件来自于千人基因组和Mills项目,里面记录了那些项目中检测到的人群Indel区域。我上面其实也提到了,候选的重比对区除了要在样本自身的比对结果中寻找之外,还应该把人群中已知的Indel区域也包含进来,而这两个是我们在重比对过程中最常用到的。这些文件你可以很方便地在GATK bundle ftp中下载,注意一定要选择和你的参考基因组对应的版本,我们这里用的是b37版本。
那么既然Indel局部重比对这么好,这么重要,似乎看起来在任何情况下都应该是必须的。然鹅,我的回答是否定的!惊讶吗!

但否定是有前提的!那就是我们后面的变异检测必须是使用GATK,而且必须使用GATK的HaplotypeCaller模块,仅当这个时候才可以减少这个Indel局部重比对的步骤。原因是GATK的HaplotypeCaller中,会对潜在的变异区域进行相同的局部重比对!但是其它的变异检测工具或者GATK的其它模块就没有这么干了!所以切记!
重新校正碱基质量值(BQSR)
在WGS分析中,变异检测是一个极度依赖测序碱基质量值的步骤。因为这个质量值是衡量我们测序出来的这个碱基到底有多正确的重要(甚至是唯一)指标。它来自于测序图像数据的base calling。因此,基本上是由测序仪和测序系统来决定的。但不幸的是,影响这个值准确性的系统性因素有很多,包括物理和化学等对测序反应的影响,甚至连仪器本身和周围环境都是其重要的影响因素。当把所有这些东西综合在一起之后,往往会发现计算出来的碱基质量值要么高于真实结果,要么低于真实结果。那么,我们到底该如何才能获得符合真实情况的碱基质量值?
BQSR(Base Quality Score Recalibration)这个步骤就是为此而存在的,这一步同样非常重要。它主要是通过机器学习的方法构建测序碱基的错误率模型,然后对这些碱基的质量值进行相应的调整。
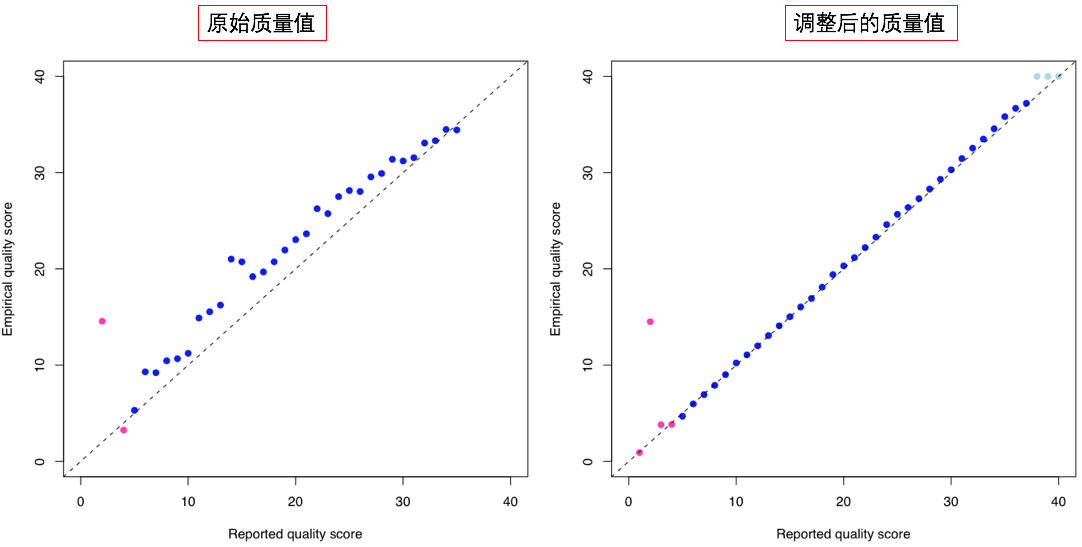
图中,横轴(Reported quality score)是测序结果在Base calling之后报告出来的质量值,也就是我们在FASTQ文件中看到的那些;纵轴(Empirical quality score)代表的是“真实情况的质量值”。
但是且慢,这个“真实情况的质量值”是怎么来的?因为实际上我们并没有办法直接测得它们啊!没错,确实没办法直接测量到,但是我们可以通过统计学的技巧获得极其接近的分布结果(因此我加了引号)。试想一下,如果我们在看到某一个碱基报告的质量值是20时,那么它的预期错误率是1%,反过来想,就等于是说如果有100个质量值都是20的碱基,那么从统计上讲它们中将只有1个是错的!做了这个等效变换之后,我们的问题就可以转变成为寻找错误碱基的数量了。
这时问题就简单多了。我们知道人与人之间的差异其实是很小的,那么在一个群体中发现的已知变异,在某个人身上也很可能是同样存在的。因此,这个时候我们可以对比对结果进行直接分析,首先排除掉所有的已知变异位点,然后计算每个(报告出来的)质量值下面有多少个碱基在比对之后与参考基因组上的碱基是不同的,这些不同碱基就被我们认为是错误的碱基,它们的数目比例反映的就是真实的碱基错误率,换算成Phred score(Phred score的定义可以参考第2节的相关内容)之后,就是纵轴的Empirical quality score了。
上面‘BQSR质量校正对比’的图中左边是原始质量值与真实质量值的比较,在这个图的例子中我们可以发现,base calling给出的质量值并没有正确地反映真实的错误率情况,测序报告出来的碱基质量值大部分被高估了,换句话说,就是错误率被低估了。
在我们的流程中,BQSR的具体执行命令如下:
- java -jar /path/to/GenomeAnalysisTK.jar \
- -T BaseRecalibrator \
- -R /path/to/human.fasta \
- -I sample_name.sorted.markdup.realign.bam \
- --knownSites /path/to/gatk/bundle/1000G_phase1.indels.b37.vcf \
- --knownSites /path/to/gatk/bundle/Mills_and_1000G_gold_standard.indels.b37.vcf \
- --knownSites /path/to/gatk/bundle/dbsnp_138.b37.vcf \
- -o sample_name.recal_data.table
- java -jar /path/to/GenomeAnalysisTK.jar \
- -T PrintReads \
- -R /path/to/human.fasta \
- -I sample_name.sorted.markdup.realign.bam \
- --BQSR sample_name.recal_data.table \
- -o sample_name.sorted.markdup.realign.BQSR.bam
这里同样包含了两个步骤:
- 第一步,BaseRecalibrator,这里计算出了所有需要进行重校正的read和特征值,然后把这些信息输出为一份校准表文件(sample_name.recal_data.table)
- 第二步,PrintReads,这一步利用第一步得到的校准表文件(sample_name.recal_data.table)重新调整原来BAM文件中的碱基质量值,并使用这个新的质量值重新输出一份新的BAM文件。
注意,因为BQSR实际上是为了(尽可能)校正测序过程中的系统性错误,因此,在执行的时候是按照不同的测序lane或者测序文库来进行的,这个时候@RG信息(BWA比对时所设置的)就显得很重要了,算法就是通过@RG中的ID来识别各个独立的测序过程,这也是我开始强调其重要性的原因。
变异检测
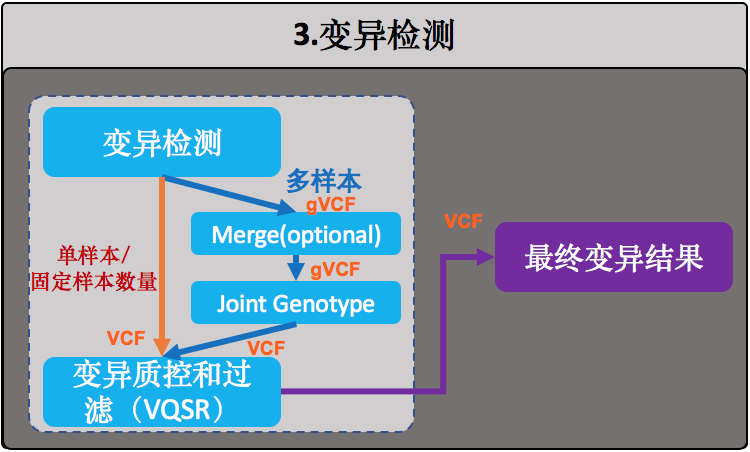
事实上,这是目前所有WGS数据分析流程的一个目标——获得样本准确的变异集合。这里变异检测的内容一般会包括:SNP、Indel,CNV和SV等,这个流程中我们只做其中最主要的两个:SNP和Indel。我们这里使用GATK HaplotypeCaller模块对样本中的变异进行检测,它也是目前最适合用于对二倍体基因组进行变异(SNP+Indel)检测的算法。
HaplotypeCaller和那些直接应用贝叶斯推断的算法有所不同,它会先推断群体的单倍体组合情况,计算各个组合的几率,然后根据这些信息再反推每个样本的基因型组合。因此它不但特别适合应用到群体的变异检测中,而且还能够依据群体的信息更好地计算每个个体的变异数据和它们的基因型组合。
一般来说,在实际的WGS流程中对HaplotypeCaller的应用有两种做法,差别只在于要不要在中间生成一个gVCF:
(1)直接进行HaplotypeCaller,这适合于单样本,或者那种固定样本数量的情况,也就是执行一次HaplotypeCaller之后就老死不相往来了。否则你会碰到仅仅只是增加一个样本就得重新运行这个HaplotypeCaller的坑爹情况(即,N+1难题),而这个时候算法需要重新去读取所有人的BAM文件,这将会是一个很费时间的痛苦过程;
(2)每个样本先各自生成gVCF,然后再进行群体joint-genotype。这其实就是GATK团队为了解决(1)中的N+1难题而设计出来的模式。gVCF全称是genome VCF,是每个样本用于变异检测的中间文件,格式类似于VCF,它把joint-genotype过程中所需的所有信息都记录在这里面,文件无论是大小还是数据量都远远小于原来的BAM文件。这样一旦新增加样本也不需要再重新去读取所有人的BAM文件了,只需为新样本生成一份gVCF,然后重新执行这个joint-genotype就行了。
我们先以第一种(直接HaplotypeCaller)做法为例子:
- java -jar /path/to/GenomeAnalysisTK.jar \
- -T HaplotypeCaller \
- -R /path/to/human.fasta \
- -I sample_name.sorted.markdup.realign.BQSR.bam \
- -D /path/to/gatk/bundle/dbsnp_138.b37.vcf \
- -stand_call_conf 50 \
- -A QualByDepth \
- -A RMSMappingQuality \
- -A MappingQualityRankSumTest \
- -A ReadPosRankSumTest \
- -A FisherStrand \
- -A StrandOddsRatio \
- -A Coverage \
- -o sample_name.HC.vcf
这里我特别提一下-D参数输入的dbSNP同样可以再GATK bundle目录中找到,这份文件汇集的是目前几乎所有的公开人群变异数据集。另外,由于我们的例子只有一个样本因此只输入一个BAM文件就可以了,如果有多个样本那么可以继续用-I参数输入:
- java -jar GenomeAnalysisTK.jar \
- -T HaplotypeCaller \
- -R reference.fasta \
- -I sample1.bam [-I sample2.bam ...] \
- ...
以上的命令是直接对全基因组做变异检测,这个过程会消耗很长的时间,通常需要几十个小时甚至几天。
然而,基因组上各个不同的染色体之间其实是可以理解为相互独立的(结构性变异除外),也就是说,为了提高效率我们可以按照染色体一条条来独立执行这个步骤,最后再把结果合并起来就好了,这样的话就能够节省很多的时间。下面我给出一个按照染色体区分的例子:
- java -jar /path/to/GenomeAnalysisTK.jar \
- -T HaplotypeCaller \
- -R /path/to/human.fasta \
- -I sample_name.sorted.markdup.realign.BQSR.bam \
- -D /path/to/gatk/bundle/dbsnp_138.b37.vcf \
- -L 1 \
- -stand_call_conf 50 \
- -A QualByDepth \
- -A RMSMappingQuality \
- -A MappingQualityRankSumTest \
- -A ReadPosRankSumTest \
- -A FisherStrand \
- -A StrandOddsRatio \
- -A Coverage \
- -o sample_name.HC.1.vcf
注意到了吗?其它参数都没任何改变,就只增加了一个 -L 参数,通过这个参数我们可以指定特定的染色体(或者基因组区域)!我们这里指定的是 1 号染色体,有些地方会写成chr1,具体看human.fasta中如何命名,与其保持一致即可。其他染色体的做法也是如此,就不再举例了。最后合并:
- java -jar /path/to/GenomeAnalysisTK.jar \
- -T CombineVariants \
- -R /path/to/human.fasta \
- --genotypemergeoption UNSORTED \
- --variant sample_name.HC.1.vcf \
- --variant sample_name.HC.2.vcf \
- ...
- --variant sample_name.HC.MT.vcf \
- -o sample_name.HC.vcf
第二种,先产生gVCF,最后再joint-genotype的做法:
- java -jar /path/to/GenomeAnalysisTK.jar \
- -T HaplotypeCaller \
- -R /path/to/human.fasta \
- -I sample_name.sorted.markdup.realign.BQSR.bam \
- --emitRefConfidence GVCF \
- -o sample_name.g.vcf
- #调用GenotypeGVCFs完成变异calling
- java -jar /path/to/GenomeAnalysisTK.jar \
- -T GenotypeGVCFs \
- -R /path/to/human.fasta \
- --variant sample_name.g.vcf \
- -o sample_name.HC.vcf
其实,就是加了–emitRefConfidence GVCF的参数。而且,假如嫌慢,同样可以按照染色体或者区域去产生一个样本的gVCF,然后在GenotypeGVCFs中把它们全部作为输入文件完成变异calling。也许你会担心同个样本被分成多份gVCF之后,是否会被当作不同的多个样本?回答是不会!因为生成gVCF文件的过程中,GATK会根据@RG信息中的SM(也就是sample name)来判断这些gVCF是否来自同一个样本,如果名字相同,那么就会被认为是同一个样本,不会产生多样本问题。
变异检测质控和过滤(VQSR)
这是我们这个流程中最后的一步了。在获得了原始的变异检测结果之后,我们还需要做的就是质控和过滤。这一步或多或少都有着一些个性化的要求,我暂时就不做太多解释吧(一旦解释恐怕同样是一篇万字长文)。只用一句话来概括,VQSR是通过构建GMM模型对好和坏的变异进行区分,从而实现对变异的质控,具体的原理暂时不展开了。
下面就直接给出例子吧:
- ## SNP Recalibrator
- java -jar /path/to/GenomeAnalysisTK.jar \
- -T VariantRecalibrator \
- -R reference.fasta \
- -input sample_name.HC.vcf \
- -resource:hapmap,known=false,training=true,truth=true,prior=15.0 /path/to/gatk/bundle/hapmap_3.3.b37.vcf \
- -resource:omini,known=false,training=true,truth=false,prior=12.0 /path/to/gatk/bundle/1000G_omni2.5.b37.vcf \
- -resource:1000G,known=false,training=true,truth=false,prior=10.0 /path/to/gatk/bundle/1000G_phase1.snps.high_confidence.b37.vcf \
- -resource:dbsnp,known=true,training=false,truth=false,prior=6.0 /path/to/gatk/bundle/dbsnp_138.b37.vcf \
- -an QD -an MQ -an MQRankSum -an ReadPosRankSum -an FS -an SOR -an DP \
- -mode SNP \
- -recalFile sample_name.HC.snps.recal \
- -tranchesFile sample_name.HC.snps.tranches \
- -rscriptFile sample_name.HC.snps.plots.R
- java -jar /path/to/GenomeAnalysisTK.jar -T ApplyRecalibration \
- -R human_g1k_v37.fasta \
- -input sample_name.HC.vcf \
- --ts_filter_level 99.5 \
- -tranchesFile sample_name.HC.snps.tranches \
- -recalFile sample_name.HC.snps.recal \
- -mode SNP \
- -o sample_name.HC.snps.VQSR.vcf
- ## Indel Recalibrator
- java -jar /path/to/GenomeAnalysisTK.jar -T VariantRecalibrator \
- -R human_g1k_v37.fasta \
- -input sample_name.HC.snps.VQSR.vcf \
- -resource:mills,known=true,training=true,truth=true,prior=12.0 /path/to/gatk/bundle/Mills_and_1000G_gold_standard.indels.b37.vcf \
- -an QD -an DP -an FS -an SOR -an ReadPosRankSum -an MQRankSum \
- -mode INDEL \
- -recalFile sample_name.HC.snps.indels.recal \
- -tranchesFile sample_name.HC.snps.indels.tranches \
- -rscriptFile sample_name.HC.snps.indels.plots.R
- java -jar /path/to/GenomeAnalysisTK.jar -T ApplyRecalibration \
- -R human_g1k_v37.fasta\
- -input sample_name.HC.snps.VQSR.vcf \
- --ts_filter_level 99.0 \
- -tranchesFile sample_name.HC.snps.indels.tranches \
- -recalFile sample_name.HC.snps.indels.recal \
- -mode INDEL \
- -o sample_name.HC.snps.indels.VQSR.vcf
最后,sample_name.HC.snps.indels.VQSR.vcf便是我们最终的变异检测结果。对于人类而言,一般来说,每个人最后检测到的变异数据大概在400万左右(包括SNP和Indel)。
这篇文章已经很长了,在变异检测的这个过程中GATK应用了很多重要的算法,包括如何构建模型、如何进行局部组装和比对、如何应用贝叶斯、PariHMM、GMM、参数训练、特征选择等等,这些只能留在后面介绍GATK的专题文章中再进行展开了。
小结
在这里,整篇文章就结束了。如你所见,文章非常长,这里基本包含了WGS最佳实践中的所有内容,但其实我想说的还远不止如此(包括CNV和SV的检测),只是暂时只能作罢了,否则恐怕就没人愿意看下去了,呵呵。在这个WGS主流程的构建过程中,我并非只是硬邦邦地告诉大家几条简单的命令就了事了,因为我认为那种做法要么是极其不负责任的,要么就是作者并非真的懂。而且如果都觉得只要懂得几条命令就可以了的话,那么我们就活该被机器和人工智能所取替,它们一定会操作得更好更高效。我想掌握工具和技术的目的是为了能够更好地发现并解决问题(包括科研和生产),所有的数据分析流程本质上是要服务于我们所要解决的问题的。
毕竟工具是死的,人是活的,需求总是会变的。理解我们所要处理的问题的本质,选择合适的工具,而不是反过来被工具所束缚,这一点很重要。个人的能力不能只是会跑一个流程,或者只是会创建流程,因为那都是一个“术”的问题,我觉得我们真正要去掌握的应该是如何分析数据的能力,如何发现问题和解决数据问题等的能力。
1F
在做局部重比对和BQSR,如果没有已知的indel数据,–knownn是否可以选择忽略而直接比对?
B1
@ stephen 你问他没用,文章抄我的,他不懂。
B2
@ 请输入您的QQ号 你是解螺旋的矿工博主?一直在关注你的微信公众号和知乎,文章写的很好,以为这篇文章作者就是你呢,所以有不理解的就直接留言了,如有疑问,该怎么向您提问呢?
2F
看完了,支持一下,学生知识有限,收藏再好好研究
B1
@ Lewis 然而,他是抄我的文章啊
B2
@ 请输入您的QQ号 抄袭可耻,支持原作者
B2
@ 请输入您的QQ号 你好,首先感谢您的分享。我想问问有没有具体讲变异检测相关的文档,主要是原理
3F
看到满满大屏全是我未经同意就被你抄过去的文章,呵呵,我好像还真没办法。
B1
@ 请输入您的QQ号 请教作者,在BQSR这一步,我试了一次,并没有达到对比图上的矫正效果,官网也说要多做几次,请问以您的经验看,进行几次比较好呀?另外如果我要进行第二轮BQSR校准,BaseRecalibrator这里的输入文件应该还是之前的bam文件还是应该用上一轮校准得到的新bam文件???多谢!!!
4F
看见评论一个劲的说抄袭,对网友的评论一概不回复,你想维权倒是把原文链接发出来啊,像你这样的抄你了你能咋地。我还真是对着这篇抄袭的文章学会操作流程了。
B1
@ 轩辕飞龙将 这都不是抄袭了,是复制粘贴啊。可能原作者的维权方式有待提高,但是你这态度,替作者叫屈啊,就像用了盗版软件出问题,还怪原创团队不维护一样。《从零开始完整学习全基因组测序(WGS)数据分析》系列文章都是复制粘贴的,http://mp.weixin.qq.com/mp/homepage?__biz=MzAxOTUxOTM0Nw==&hid=1&sn=d945cf61bd86e85724e146df42af5bcc&scene=18#wechat_redirect
B2
@ 无可奈何花落去 我只是在说原作者维权方式不对,我想看原文都看不了我能咋办
来自外部的引用