

Tophat+cufflinks组合是RNA-Seq数据分析的一个很经典的分析方法了,四年前关于这两个软件的使用,Nature Protocol专门发文介绍如何使用这两个软件,具体可以参考《利用tophat和Cufflinks做转录组差异表达分析的步骤详解》。前段时间PLoB上给大家推荐了大牛Steven L. Salzberg实验室新开发的三个用于RNA-Seq转录组数据分析的新工具HISAT、StringTie和Ballgown,具体请阅读《RNA-Seq分析新工具》。同时Steven L. Salzberg实验推荐大家用这三个软件来代替之前的Tophat+cufflinks。为了方便大家使用HISAT、StringTie和Ballgown,Nature Protocol最近良心套餐又出来了。新的一篇文章专门介绍了这三个软件的使用。文章摘要和链接如下,文章全文已经放在PLoB生物信息学群共享文件。
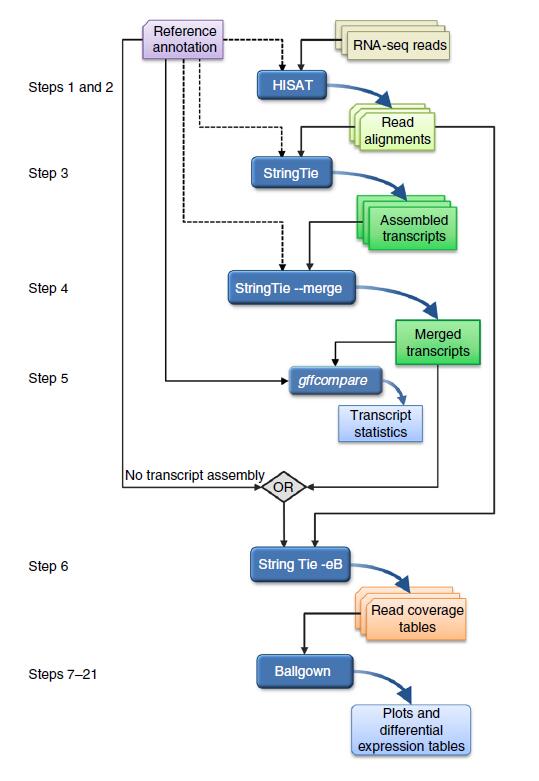
Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown。
Mihaela Pertea, Daehwan Kim, Geo M Pertea, Jeffrey T Leek & Steven L Salzberg
High-throughput sequencing of mRNA (RNA-seq) has become the standard method for measuring and comparing the levels of gene expression in a wide variety of species and conditions. RNA-seq experiments generate very large, complex data sets that demand fast, accurate and flexible software to reduce the raw read data to comprehensible results. HISAT (hierarchical indexing for spliced alignment of transcripts), StringTie and Ballgown are free, open-source software tools for comprehensive analysis of RNA-seq experiments. Together, they allow scientists to align reads to a genome, assemble transcripts including novel splice variants, compute the abundance of these transcripts in each sample and compare experiments to identify differentially expressed genes and transcripts. This protocol describes all the steps necessary to process a large set of raw sequencing reads and create lists of gene transcripts, expression levels, and differentially expressed genes and transcripts. The protocol's execution time depends on the computing resources, but it typically takes under 45 min of computer time. HISAT, StringTie and Ballgown are available from http://ccb.jhu.edu/software.shtml.
1F
恩