

1. speedseq的安装
1.1 安装前先需安装好python (建议升级到2.7最新版2.7.13),而且需要安装pysam库(需升级到最新版,这里升级到0.11.2版本),如果没有升级,可能在speedseq sv方法中报如下错:
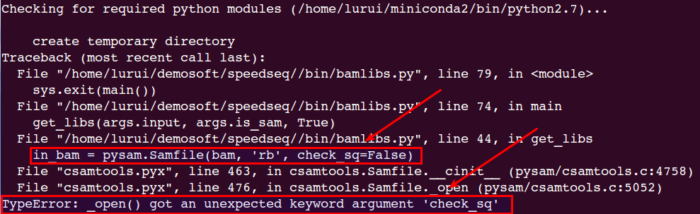
1.2 再安装root,只有安装了它cnvnator才可以正常安装。
从官网〔1〕下载root,有Binary distributions但是需要注意下载对应系统的版本(例如我这里下载的Ubuntu 14版本的)。
- wget https://root.cern.ch/download/root_v6.10.08.Linux-ubuntu14-x86_64-gcc4.8.tar.gz
- tar -zxvf root_v6.10.08.Linux-ubuntu14-x86_64-gcc4.8.tar.gz
# 解压为文件夹root
再修改\$HOME/.bashrc,添加以下两行
- export PATH=/Path/to/rootdir/bin:$PATH
- export LD_LIBRARY_PATH=/Path/to/rootdir/lib:$LD_LIBRARY_PATH
注意:/Path/to/rootdir即上一步安装root包时压缩后root文件夹的位置,请用绝对路径!
如果后期运行cnvnator时报错如下,则表明此处LD_LIBRARY_PATH设置有误。
error while loading shared libraries: libCore.so: cannot open sh
〔出现这种报错,可查看http://blog.sciencenet.cn/blog-653020-626976.html〕
再运行以下代码
- cd root
- . bin/thisroot.sh
以上安装过程参考[2]
再按照官网[3]上的步骤下载安装speedseq
- git clone --recursive https://github.com/hall-lab/speedseq
- cd speedseq
- make
# 经测试gcc 4.4.7版安装会有报错,gcc 4.8.5可正常安装
1.3 如果想用VEP做注释,还需先手动安装
正确安装方法如下:
- curl -OL https://github.com/Ensembl/ensembl-tools/archive/release/76.zip
- SPEEDSEQ_DIR=$PWD
# 注意:这里是speedseq安装目录(也是我的当前目录),以下操作我都在speedseq目录下完成!
- unzip 76.zip
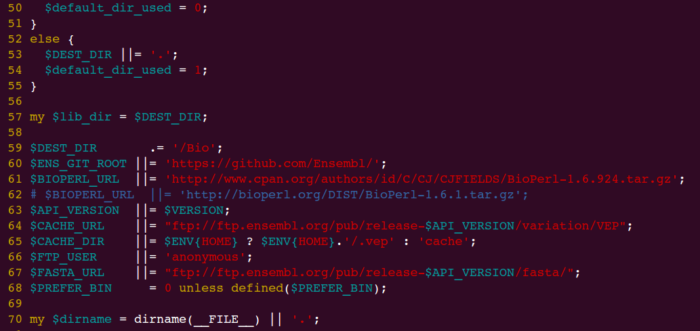
注意:因为bioperl下载地址改变,按照speedseq官网上安装会报错!一定要修改INSTALL.pl再运行。
- vim ensembl-tools-release-76/scripts/variant_effect_predictor/INSTALL.pl
将第61行的BIOPERL_URL修改为以下地址,INSTALL.pl中的地址已失效,
http://www.cpan.org/authors/id/C/CJ/CJFIELDS/BioPerl-1.6.924.tar.gz
截图如下:
如果你研究的是人,可以用以下代码在本地建立人的数据库
- perl ensembl-tools-release-76/scripts/variant_effect_predictor/INSTALL.pl \
- -c $SPEEDSEQ_DIR/annotations/vep_cache \
- -a ac -s homo_sapiens -y GRCh38
# 它会自动下载人的全基因组序列,具体地址如下:ftp://ftp.ensembl.org/pub/release-76/variation/VEP/homo_sapiens_vep_76_GRCh38.tar.gz,大小约3.1G
- cp ensembl-tools-release-76/scripts/variant_effect_predictor/variant_effect_predictor.pl $SPEEDSEQ_DIR/bin
- cp -r Bio $SPEEDSEQ_DIR/bin
speedseq非常赞的原因之一是它几乎集成了所有需要的三方软件(注:其中有些比较难安装),而且在/Path/to/speedseq/bin/目录下有个speedseq.config,其中记录了所有三方软件可执行程序的路径。这样就可以完全独立了(比方你系统环境变量中有多个samtools,或者多个python,你想用哪一个,就可以直接在这里指定)
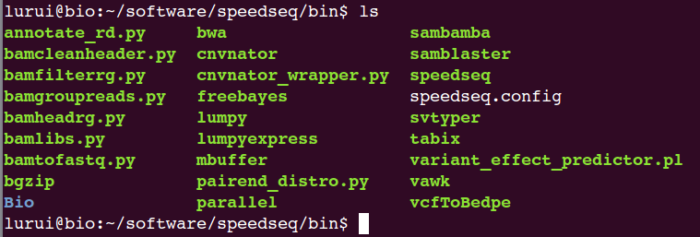
安装完后进行测试,查看是否安装完整,建议将speedseq.config中每一行都测一下,看路径中的程序是否能正常运行。
我的/Path/to/speedseq/bin/下的程序如下:
2. speedseq的使用
像所有的好软件一样,speedseq也提供了example,在/Path/to/speedseq/example/目录下,该目录下有一个data目录(注意:data目录中的reference是bwa index过的)。
建议按/Path/to/speedseq/bin/run_speedseq.sh中代码一句句运行
2.1 speedseq align
注意:speedseq调用bwa-mem后,还生成了discordants.bam和splitters.bam,这两个文件是lumpyexpress必须的(这个已整合在speedseq中),那么这两个bam是怎么来的?
按照lumpy github官网[4]上的描述
- speedseq align -R "@RG\tID:id\tSM:sample\tLB:lib" \
- human_g1k_v37.fasta \
- sample.1.fq \
- sample.2.fq
注意:使用speedseq align时建议加上-o选项,它指定输出文件(夹)前缀;-t选项,它指定线程数(默认单线程)
以上等价于
- # Align the data
- bwa mem -R "@RG\tID:id\tSM:sample\tLB:lib" human_g1k_v37.fasta sample.1.fq sample.2.fq | samblaster --excludeDups --addMateTags --maxSplitCount 2 --minNonOverlap 20 | samtools view -S -b - > sample.bam
- # Extract the discordant paired-end alignments.
- samtools view -b -F 1294 sample.bam > sample.discordants.unsorted.bam
- # Extract the split-read alignments
- samtools view -h sample.bam \
- | scripts/extractSplitReads_BwaMem -i stdin \
- | samtools view -Sb - \
- > sample.splitters.unsorted.bam
- # Sort both alignments
- samtools sort sample.discordants.unsorted.bam sample.discordants
- samtools sort sample.splitters.unsorted.bam sample.splitters
2.2 speedseq var
这里调用的是freebayes,它的精确度与GATK不相伯仲,具体可能查看speedseq的引文[5]。
speedseq var除例子中的-o选项(指定输出文件前缀)外,还有以下几个选项值得注意:
-q 指定最低变异质量值(质量值小于这个值的将认为是不可信结果)
-A 使用VEP对变异检测结果进行注释,当然前提是安装好了vep,speedseq/bin中variant_effect_predictor.pl可用
2.3 speedseq的sv方法
建议加上以下选项
-g表示使用svtyper对SV breakends进行分型
-d表示使用CNVnator来计算read深度
-P 在vcf中输出LUMPY的概率曲线
参考材料
[1] https://root.cern.ch/content/release-61008
[2] http://www.cpp.edu/~pbsiegel/phy499/rootinstall.html
[3] https://github.com/hall-lab/speedseq
[4] https://github.com/arq5x/lumpy-sv
[5] chiang C., Layer RM., Faust GG, et al. SpeedSeq: ultra-fast personal genome analysis and interpretation.Nature Methods. 2015 Oct;12(10):966-968