

小伙伴们,大家好,今天我们来开启一个新的话题,Single cell sequence,近来单细胞测序在探索生物过程、疾病机理等方面展现了前所未有的精度,通过对单细胞进行 DNA 和 RNAseq 我们得以高精度的了解单个细胞水平的体细胞突变情况,并且对于一个样本中细胞类型的组成和解析获得空前的认识。
那么,我们如何开展单细胞测序?单细胞测序的原理是怎样的那?我们如何开展单细胞测序? 单细胞测序的数据处理与一般的 RNA-seq、DNA-seq 有何不同?
第一讲
好的,言归正传,首先我们来看看如何获得单细胞,从而进行下一步的测序过程。
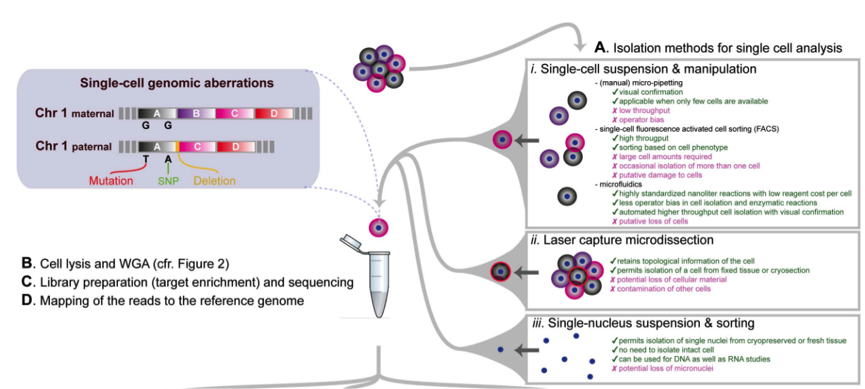
为了进行单细胞测序实验,首先我们需要分离出感兴趣的单细胞,比较常用的方法之一是使用流式细胞仪(FACS)方法进行分离,我们可以使用无限制的分选技术,从而可以获得随机样品,同时也可以选择散射性质等设置 sorting gate。不仅如此,我们还可以使用荧光标记的抗体获得细胞表面特异 marker 的细胞亚群,从而探究这一 sub group 细胞的特性。
目前的技术允许同时检测多达 20 个参数,因此可以将单细胞分选到 96 或 384 孔板中进行接下来的单细胞测序。但是 FACS 也有缺点,首先需要较大的起始细胞量,同时可能导致有些孔中有两个,有些孔中没有细胞。
显微操作技术提供了获得少量细胞的替代方法,我们可以通过直接特定位置摘取单个细胞进行测序。但是这种方法依赖于人工操作,无法实现高通量进行。
最近,微流控技术在单细胞分选上展现良好的应用,微流控技术可以对整个过程进行良好的监控和处理。微流控的缺点主要在于固定的微流控芯片等。
大家可以通过观察上图来看到每种方法的优缺点。
在获得单细胞之后,对于 single cell DNA-seq,我们该如何开展下面的实验那?
在多细胞生物体中,由于细胞存在着有丝分裂,在传代过程中 DNA 复制存在着各种各样的突变,这些突变通过积累最终可以导致癌症等多种疾病的方法,在生殖细胞发育过程中,也可以看到不同种基因组突变,包括替换、插入和删除、拷贝数变异、染色体结构重组等。这些过程,我们可以通过 single cell DNA-Seq 来了解,但是对于一个细胞而言,一个哺乳动物细胞,通常的 DNA 只有不到 10 pg,因此我们首先需要对全基因组进行扩增才能进行下一步的实验。
目前的全基因组扩增技术(WGA)主要是通过 PCR、多重替换扩增技术(MDA)或两者的组 合来实现。
PCR 策略主要是通过使用随机引物进行扩增,然后引入通用 adaptor。

MDA 方法则利用具有链置换活性的 DNA 聚合酶,∅29 进行恒温扩增,这是一种滚环复制酶, 可以处理长达 10 Kb 的序列,一旦接触到前面结合的序列后,聚合酶可以释放前者 DNA,从而释放用于进一步的扩增。对于这两种方法,随机引物 PCR 方法实验更高水平的一致扩增, 但是覆盖度较低。MDA 方法由于 DNA 聚合酶的高保真性和高处理特性,可以实现较好的覆盖度也有利于 SNP 等精细实验,然而由于 GC 含量等影响,MDA 可能导致不一致扩增,Gcbias 比较大。因此 MDA 用于 CNV 检测等的假阳性概率比较大。华人科学家谢晓亮等人基于前两种方法提出综合两种方法的 MALBAC 方法,实现了高覆盖度的一致性扩增方法。
通过增加互补末端引物使用滚换复制酶产生可以末端配对的 loop,被扩增出来的 DNA,由于末端配对,因此无法被称为接下来的模板,从而可以实现近乎线性的扩增,经过五轮线性扩增后,然后通过 PCR 进行指数级扩增,相对于 MDA 方法 31%-61% 的丢失率,MALBAC 方法只有 1% 的缺失覆盖率。
同时对于 SNP 和 CNV 的检测的假阳性更低。
其他的方法用于减少扩增时的不一致性则主要是通过减少反应体系,通过使用 nano-liter 级别的反应体系,增加有效的模板浓度,这类技术被称为 MIDAS 技术。

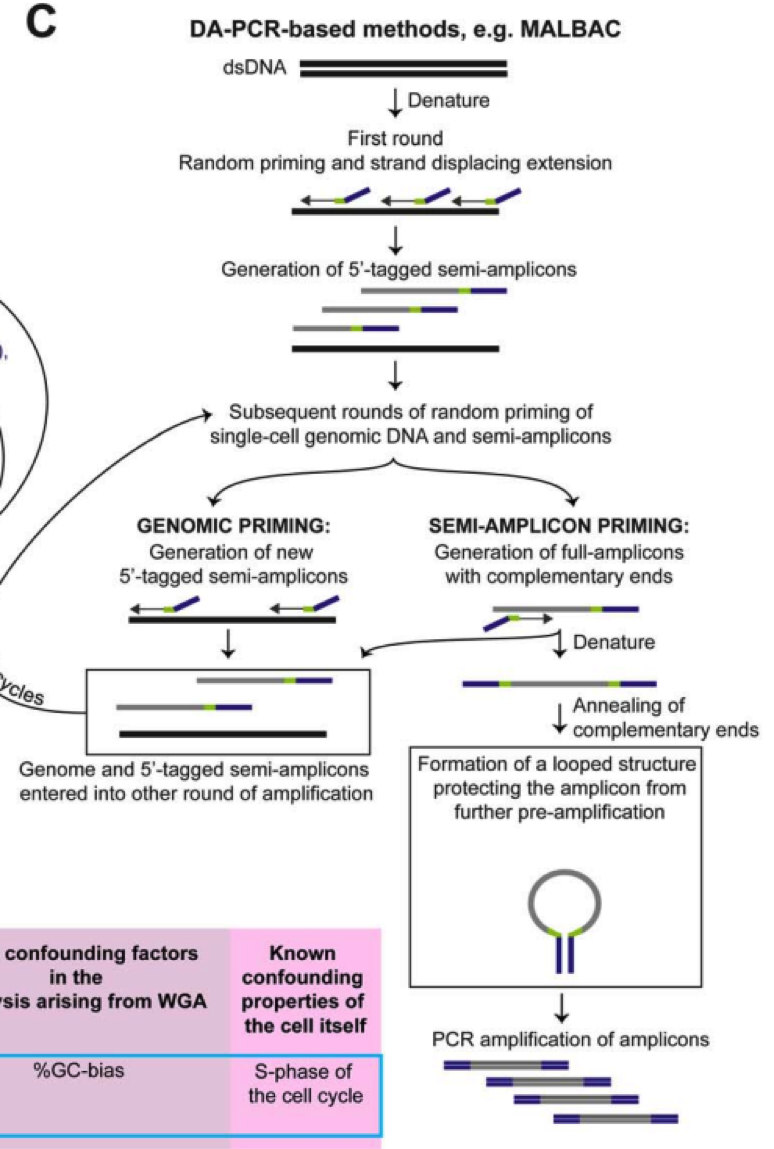
这里有一份总结各类技术主要是使用方法的表格,方便大家使用。
在获得单细胞基因组测序之后,首先我们需要监控 read 的质量剔除低质量的 reads,然后进行基因组的 map 工作等,通过矫正由于 GC 含量引起的误差之后,我们可以多种算法 breakpoint,CNV 等。
这里向大家推荐 GATK 工具包,这个工具包包含了处理下一代测序和virant calling的线管技术。
学会了 DNA-seq 的初级处理之后,我们如何对不同的细胞之间进行比较和聚类?进行详细的数据挖掘?我们如何进行 RNA-seq?
第二讲
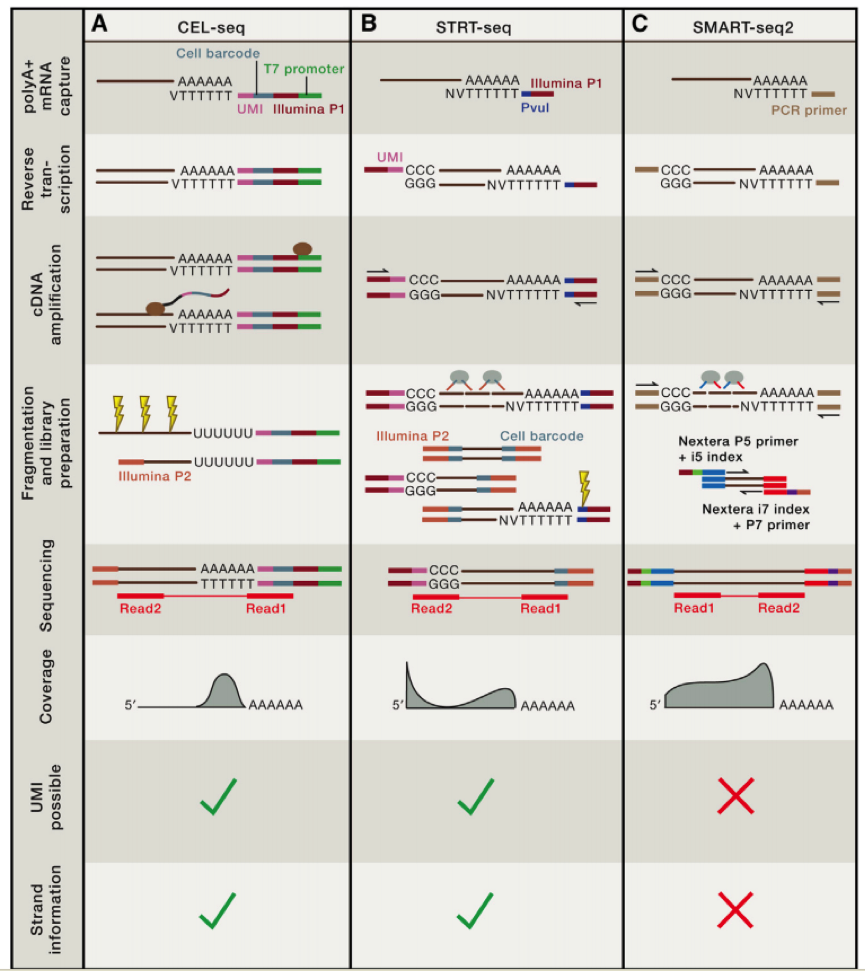
上一讲我们讲到了不同单细胞 DNA 测序的方法,比较了不同测序方法的异同?大家还记得吗?这里给一张图片供大家复习参考:
今天,我们主要来了解一下单细胞 RNA-seq 的方法,使用 microarray 和 RNA-seq 我们可以获得在不同组织和细胞中基因表达的强弱,显而易见,单细胞 RNA-seq 方法为我们具体一个组织内,或者不同类型细胞的表达差异,同时还可以为我们发现新的细胞类型奠定基础,单细胞 RNA-seq 的主要障碍是如何从单个细胞中获取充分量的 RNA 用于下一步的测序,不同的方法被开发出来用于扩增低于 pg 级的 mRNA。
这些方法的主要障碍是扩增的 bias 问题,对于来自不同基因的 mRNA 之间的差异比较产生干扰。
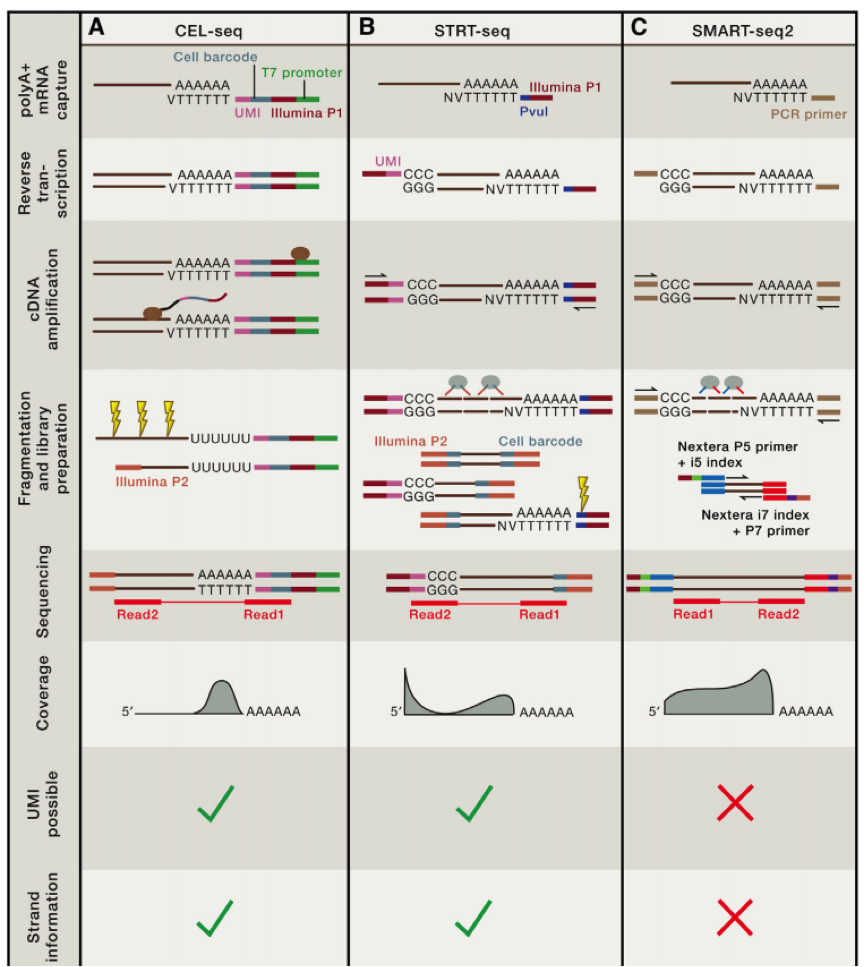
第一个利用单细胞进行 RNA-seq 是 Surani 实验室在 2009 年开发出来的,第一作者汤富酬大神,自此 Tang 大神在 single cell 领域风生水起,这是后话,接下来人们用 single cell RNA-Seq 跟踪 MES cell 的发育分化情况,扩增的方法主要是使用将 RNA pull down 下来,然后使用 polyT 引物反转录带 ployA 的 RNA,同时引物上含有特定的 anchor 序列。

科学家们在借鉴了基于 microarray 用于 single cell sequence 的技术后,修改了一些实验条件,比如延伸时间等,从而获得全长的 cDNA,进一步通过 SOLID 测序,我们可以看到具有非常好的稳定性。
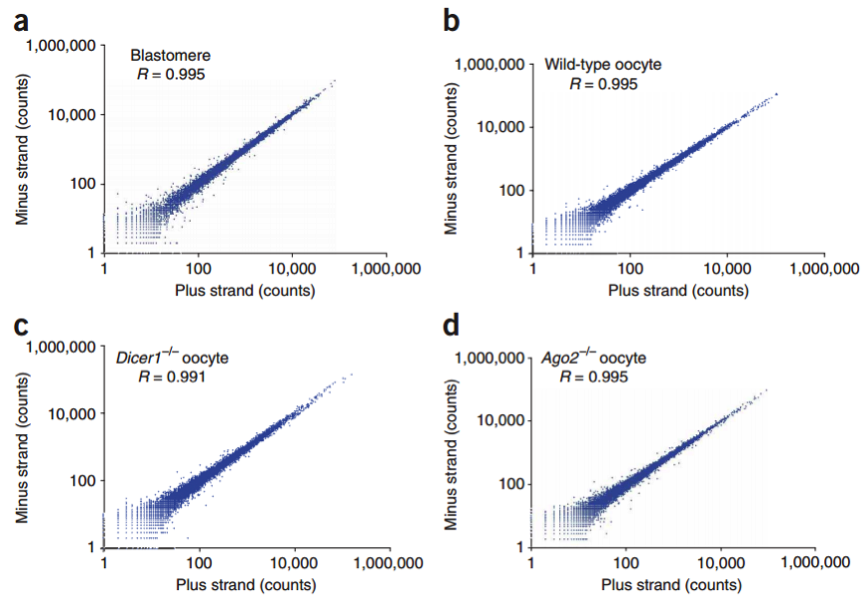
在这个技术之后,2011 年科学家们使用了新的 cell barcode 方法,从而可以复杂的探究多个复杂细胞的特性,称为 STRT 技术,但是 STRT 技术对于 5’end 具有偏好性,基于转座子酶,人们开发了升级版的 STRT2 技术。
为了减少由于扩增引起的误差,人们在一些单细胞测序的步骤中增加了 UMI(unique molecular identifiers),UMIs 是由 4-10 个随机核苷酸组成的序列,在 mRNA 反转录后,进入到文库中,每一个 mRNA,随机连上一个 UMI,因此可以计数不同的 UMI,最终计数 mRNA 的数量。
不同的方法的优劣我们可以在最上面的图中得到,那么我们如何进行选择哪?
如果我们想了解不同细胞间的异质性,那么我们需要减少实验的 bias,推荐使用 UMI 相应的技术,如果我们想了解 RNA 全长,包括 splice site 等信息,那么我们就需要选择包含全部转录长度的技术。
目前多种技术可以通过多通道微流控自动处理技术进行处理,可以允许自动分离和处理细胞,同时控制细胞的数量,实验的误差,我们相信随着实验技术的改进,single cell sequence 终有一日,会成为常用的实验技术,助力人类了解自己,了解生物!!
第三讲
学完前两讲后,有没有对于单细胞测序有一个初步的认识那?这一讲我们主要来聊一聊如何利用单细胞 RNA-seq 数据,进行数据挖掘?
首先让我们来考虑一个非常关键性的问题,当我们如果想做单细胞测序,那么我们应该如何设计我们的实验?
说的更直白一些,我们需要测多少细胞?测多深?
单细胞 RNA-seq 为我们了解组织的组成等提供非常好的工具,而单细胞测序的效果也取决于我们到底测多少细胞,一般而言几百个细胞的单细胞测序不仅仅捕获常见的细胞成分,对于一些稀少的细胞类型也会测到,不过,需要考虑的情况是我们捕获细胞的方法是否具备一定的偏颇?比如不同类型细胞细胞的大小?细胞的形态?是否会对细胞的获取有所倾向?同时我们还需要实验产生的误差,我们需要估计我们所操作的成功率,由于 RNA 降解,细胞的凋亡等,这些数据将会被排除,我们初始选择的细胞个数往往会大于我们目标测序的细胞个数。
那么第二个问题,我们需要测多深?
如果我们使用 UMI 计数方法( UMI 计数,是指在建库过程中,通过在引物上,增加随机序列,则对于同一种 mRNA 连上同样的 UMI 概率几乎为 0,则我们可以忽略由于 PCR 造成的误差,对于一种 mRNA,测到的 UMI 数量可以近似看成 mRNA 的表达个数),推荐至少每一个转录本至少覆盖 3-4 倍,确保一些低表达的 mRNA 不会因为 noise 而被忽略,一般而言,illumina 的一条 lane 中最终可以被使用的数据大概占 50%,另一些 reads 因为实验技术所限,为一些 adaptor 等无效序列。
这里我们做一个计算题:
一个细胞假设我们最终想得到大概 10000 种转录本的信息,那么对于一个 lane 而言( nextseq 机器可以有 200million 条 reads ),那么可以测多少细胞那?200,000,000/10000/4/2=2500 大概可以测 2500 个细胞!好了,那我们假设如果我们获得了数据后,那我们该如何进行下一步的数据分析那?

首先我们需要对测序的质量进行评估,截去低质量部分,这里推荐的工具为 Fastqc,以往的文章中,大程跟大家介绍过 fastqc,还记得不?
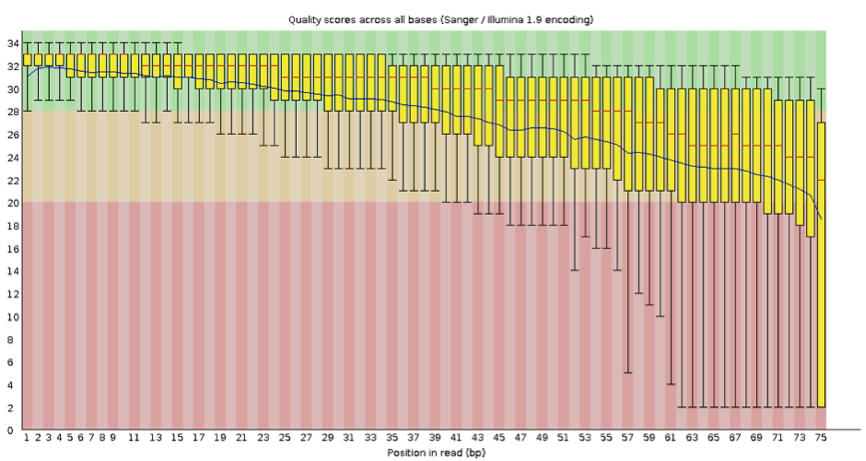
这里小编推荐大家最终截取的长度需要大于 35bp,保证匹配的真实性,对质量进行评估后,我们需要进行匹配,常用的包括 BWA,bowtie 等,好了问题来了,我们选择什么作为 reference?
如果我们选择基因组作为 reference,那么一个显著的问题是可能匹配到假基因等位置,影响我们的定量评估。
使用转录组作为参考可以显著的减少比对空间,考虑到不同的剪切问题,如果我们对于不同转录本,没有要求的话,推荐大家使用多转录本 merge 的序列。注意了,注意了,注意了!!
因为大部分单细胞测序主要是富集 3‘UTR 部分,那么如果我们使用 cufflink,就会造成很大的问题,当我们比对完后,需要确定每个转录本的表达量,前面我们说到UMI用于 RNA 的计数,在计数前,我们还需要对不同的 cell 进行滤过。
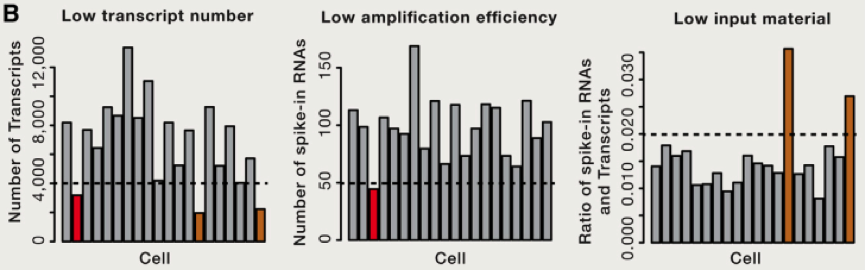
由于存在 RNA 降解等多种问题,这里我们需要考虑到去除一些低转录本等情况,同时我们还需要考虑去除低扩增效率的 cell 等。那么我们如何比较不同 cell 之间的差异那?首先需要做的便是归一化,这里跟大家介绍使用 spike-in RNA 进行归一化的方法,spike-in RNA 往往是我们在建库过程中使用的人为计数后,掺入的 RNA,可以帮助我们对不同 cell 之间的进行横向的比对,如果我们没有 spike-in 怎么办呐?也不必害怕,往往简单的将总共转录本进行计数,也是可以的。我们可以将所有的 cell 归一化到 reads 总数是中位数的细胞的 reads 总数中,或者将所有细胞归结成同样的 reads,被称为 down-sampling。
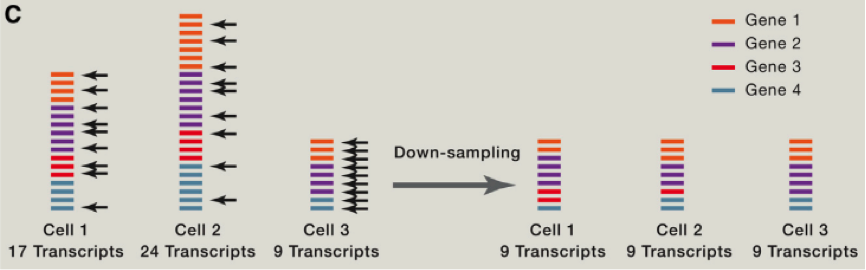
在获得不同细胞不同基因的表达后?我们该如何从中挖掘出我们想要的信息那?首先如果我们对一个组织或者器官等进行单细胞测序,我们首先能做的便是分析其中可能的细胞亚类。
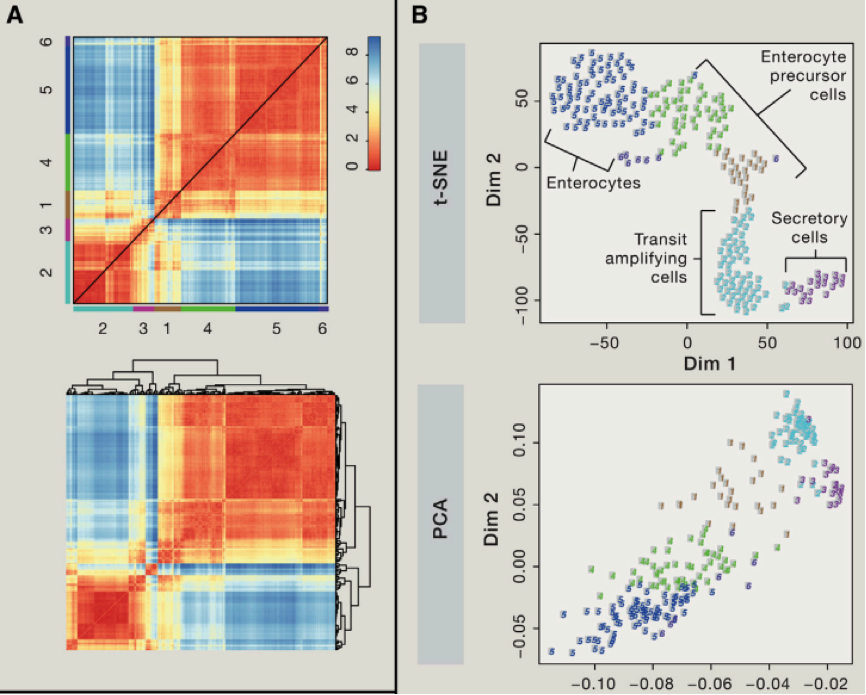
我们可以使用主成分分析(PCA)等,对细胞群进行聚类分析,我们可以利用第一主成分,第二主成分等对细胞进行分类,如果我们想要了解精细的细胞类别,我们可以结合使用层次聚类方法。不过我们需要考虑由于 batch effect 造成的假阳性聚类问题等。另外还需要考虑由于细胞本身,比如细胞处于不同细胞周期造成的异质性,当然我们可以通过微流控技术在最开始的过程中,就减少可能存在的复制中的细胞,另外我们也可以对于 cell-cycle related 基因变化进行剔除。
聚类中,一个比较难的地方,在于如何鉴定一些稀少的细胞类型,对于一些占比重 1% 以下的细胞,该如何进行聚类那?,
近期有一个课题组通过首先使用 k-means 聚类方法寻找常见的细胞类型,进而对于 outlier 进行 screen 寻找。
这个方法(RaceID)以高敏感性寻找到了一些在肠道中稀有的细胞群落。
那么我们找到这些不同类型的细胞类型后,我们该干什么那?
一个很关键的问题,我们需要鉴定一些 marker gene,比如我们可以使用差异基因分析工具,诸如 DESeq 等来推测相关 marker 基因知道下游的分析。
大家有没有听懂那?
参考文献:

Tang, F., Barbacioru, C., Wang, Y., Nordman, E., Lee, C., Xu, N., Wang, X.,Bodeau, J., Tuch, B.B., Siddiqui, A., et al. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods6, 377–382.
1F
您好,您能否将本篇教程的所涉及所有的reference发给我一下,或者将list发给我?