

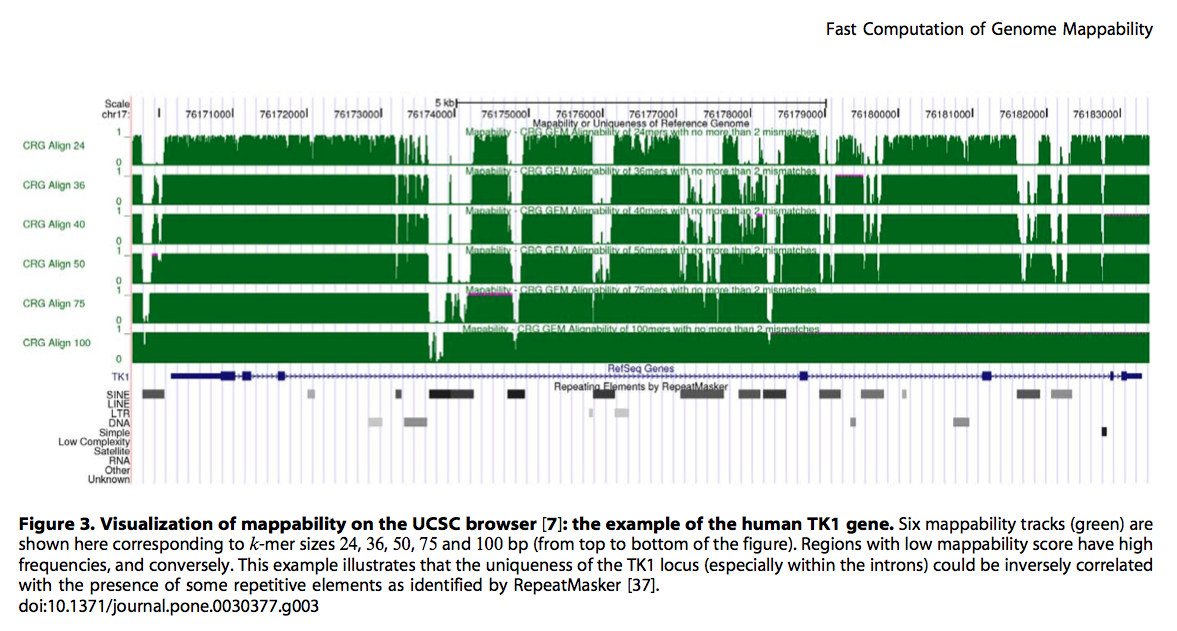
现在测序应用越来越广泛,各种Seq满天飞,譬如ChIP-Seq, DNase-Seq等等。在align完测序的reads后,有些位置reads多有些位置reads 少。除了本身的生物特性外是否还与其他的因素有关呢?
当然最主要的因素应当是基因组序列本身,毕竟基因组不是简单的ACGT的完全随机。另外一个因素就是reads的长度。今天给大家介绍利用GEM-library(论文Derrien et al in 2012 [1],软件下载 [2])创建基因组Mappability文件。这个就是基于基因组本身创建出在特定条件下(譬如reads长度50bp,允许2个mismatch)基因组各位置unique reads的覆盖情况。
下面主要利用https://wiki.bits.vib.be/index.php/Create_a_mappability_track#cite_note-2 网站的基本方法来给大家介绍一下如何以酵母基因sacCer3为例生成Mappability文件。
1.下载参考数据
1.1 Download the yeast reference genome data from the UCSC table repository
- UCSC data can be obtained from their FTP server http://hgdownload.soe.ucsc.edu/goldenPath/sacCer3[3]
- other files are built from the table browser with interaction in your browser
We use below Kent tools (unix version from [4], also available for macOSX[5]) used by the UCSC team to produce the Genome browser system. Some of these tools should be present on your machine in order to repeat the code below.
- # create a folder to store all results
- basefolder="~/Desktop/alignability/sacCer3_alignability"
- mkdir –p ${basefolder} && cd ${basefolder}
- # download the 2bit file
- wget http://hgdownload.soe.ucsc.edu/goldenPath/sacCer3/bigZips/sacCer3.2bit
- # extract the fasta data with Kent\'s \'twoBitToFa\'
- twoBitToFa sacCer3.2bit SacCer3.fa
- # also create a two-column list of chromosomes names and sizes with Kent\'s \'faSize\'
- faSize sacCer3.fa -detailed > sacCer3.sizes
1.2 download additional SacCer3 annotations
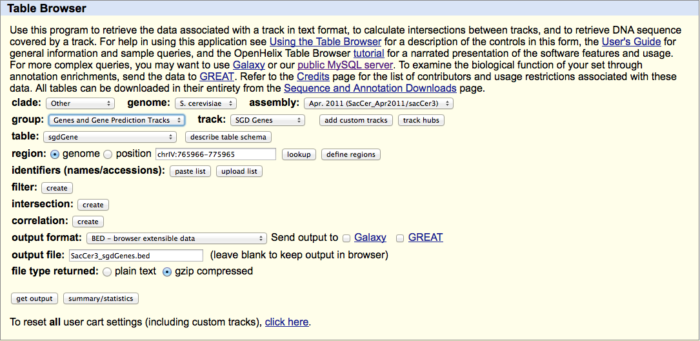
Get the SGD gene table as well as the GC distribution across the whole genome from the UCSC-table (http://genome.ucsc.edu/cgi-bin/hgTables[6]).
注意:the chromosome names should be identical between the 2bit-fasta and the gene table in order for IGV to display them together
- # create a folder to store all results
- basefolder="~/Desktop/alignability/sacCer3_alignability"
- mkdir –p ${basefolder} && cd ${basefolder}
- # get the sgdGene table for other purposes (this is not a BED file)
- wget http://hgdownload.soe.ucsc.edu/goldenPath/sacCer3/database/sgdGene.txt.gz
- # also get it in the BED-format \' SacCer3_sgdGenes.bed\' using the Table browser (see screen-shot below)
- # get and adapt GC% data in variable steps
- wget http://hgdownload.soe.ucsc.edu/goldenPath/sacCer3/gc5Base/sacCer3.gc5Base.varStep.txt.gz
- # the file: sacCer3.gc5Base.varStep.txt.gz is the raw data used to encode
- # the gc5Base track on sacCer3. This file was produced from the sacCer3.2bit
- # sequence with the kent source tree utility hgGcPercent (see below)
1.3 create a 5base step file from the variable step with Kent\'s hgGcPercent
We can produce the GC% wig file directly from the 2bit genome and name it slightly differently to the downloaded version.
- # create a folder to store all results
- basefolder="~/Desktop/alignability/sacCer3_alignability"
- mkdir –p ${basefolder} && cd ${basefolder}
- hgGcPercent -wigOut \
- -doGaps \
- -file=stdout \
- -win=5 \
- sacCer3 \
- sacCer3.2bit | \
- gzip > sacCer3.gc5Base.varStep.wig.gz
1.4 create a bigwig version for rapid visualization
The wigToBigWig command is somehow sensitive to complex fasta headers. If you Fasta sequences have long names including spaces and \':\', you should better simplify it and keep only one word like \'1\' or \'chr1\' to avoid problems at this stage
- # create a folder to store all results
- basefolder="~/Desktop/alignability/sacCer3_alignability"
- mkdir –p ${basefolder} && cd ${basefolder}
- wigToBigWig <(bgzip -cd sacCer3.gc5Base.varStep.wig.gz) \
- sacCer3.sizes sacCer3.gc5Base.bw
2 Install and run the GEM library tools
Tools were obtained and installed as described in http://algorithms.cnag.cat/wiki/GEM:Installation_instructions[7] (the core-2 version GEM-binaries-Linux-x86_64-core_2-20130406-045632.tbz2 was used on the local machine!)
there is not yet a version for apple OSX and the source is not available to build locally
2.1 create a \'gem\' index
- # create a folder to store all results
- basefolder="~/Desktop/alignability/sacCer3_alignability"
- mkdir –p ${basefolder} && cd ${basefolder}
- pref="sacCer3"
- reference="sacCer3.fa"
- idxpref="sacCer3_index"
- thr=8; # use 8 cores
- gem-indexer -T ${thr} -c dna -i ${reference} -o ${idxpref}
- #结果文件
- -rw-rw-r-- 1 splaisan splaisan 35M Dec 18 14:15 SacCer3_index.gem
- -rw-rw-r-- 1 splaisan splaisan 1.9K Dec 18 14:15 SacCer3_index.log
2.2 compute Alignability for several K-mer lengths
The created index can now be used to compute mapability across the full genome. The alignability / mappability is computed for kmers between 50 and 250 bases by steps of 50 bases and the results are immediately converted to WIG and BigWIG format for IGV visualization.
2.3 create for 5 different read sizes
We selected increasing sizes corresponding to read material available on public repositories like SRA and showing the effect of read size on the uniqueness of subsequent short read alignment.
our tracks are based on single-end reads, paired reads are obviously more easily uniquely mapped due to their increased span size that can reach-out from difficult regions
The wigToBigWig command is somehow sensitive to complex fasta headers. If you Fasta sequences have long names including spaces and \':\', you should better simplify it and keep only one word like \'1\' or \'chr1\' to avoid problems at this stage
- # create a folder to store all results
- basefolder="~/Desktop/alignability/sacCer3_alignability"
- mkdir –p ${basefolder} && cd ${basefolder}
- pref="sacCer3"
- idxpref="sacCer3_index"
- thr=8; # use 8 cores
- for kmer in $(seq 50 50 250); do
- # compute mappability data
- gem-mappability -T ${thr} -I ${idxpref}.gem -l ${kmer} -o ${pref}_${kmer}
- # convert results to wig and bigwig
- gem-2-wig -I ${idxpref}.gem -i ${pref}_${kmer}.mappability -o ${pref}_${kmer}
- wigToBigWig ${pref}_${kmer}.wig ${pref}.sizes ${pref}_${kmer}.bw
- done
说明:
在利用gem-mappability生成文件的时候reads长度一般可以选择36,59,75,100。这些是目前illumina机器测序的主要长度。实际应用可以按照自己的需要来选择参数-l。
3. Visualize results in IGV
A screen-shot is provided to illustrate the results using IGV and the Broad sacCer3 loaded genome. Other yeast genomes available in IGV include:
Other strains are available from:
- http://genomevolution.org/wiki/index.php/SGRP:_Sanger_Institute_Yeast_Genomes[8]
- (e.g. Y55) http://genomevolution.org/CoGe/services/JBrowse/service.pl/sequence/9136[9]
Reads for yeast genome can be obtained from the Sanger institute at:
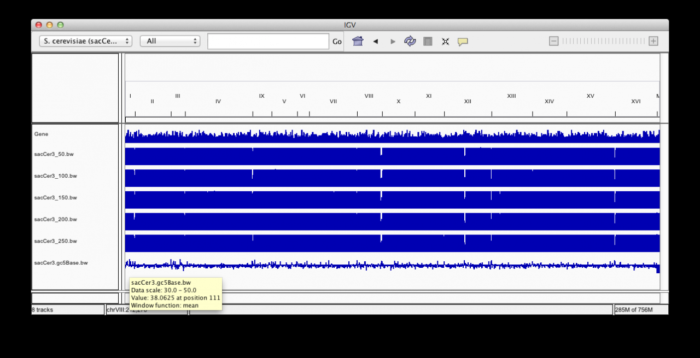
Legend:
- top track represents gene density across the genome for SGD genes
- next 5 tacks show alignability / mappability for increasing kmer length.
- bottom track shows GC% across the genome in 5base bins (scale set between 30 and 50)
参考:
- Thomas Derrien, Jordi Estellé, Santiago Marco Sola, David G Knowles, Emanuele Raineri, Roderic Guigó, Paolo Ribeca . Fast computation and applications of genome mappability.
- http://algorithms.cnag.cat/wiki/The_GEM_library
- http://hgdownload.soe.ucsc.edu/goldenPath/sacCer3
- http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64
- http://hgdownload.cse.ucsc.edu/admin/exe/macOSX.i386"
- http://genome.ucsc.edu/cgi-bin/hgTables
- http://algorithms.cnag.cat/wiki/GEM:Installation_instructions
- http://genomevolution.org/wiki/index.php/SGRP:_Sanger_Institute_Yeast_Genomes
- http://genomevolution.org/CoGe/services/JBrowse/service.pl/sequence/9136
- http://www.sanger.ac.uk/research/projects/genomeinformatics/sgrp.html