

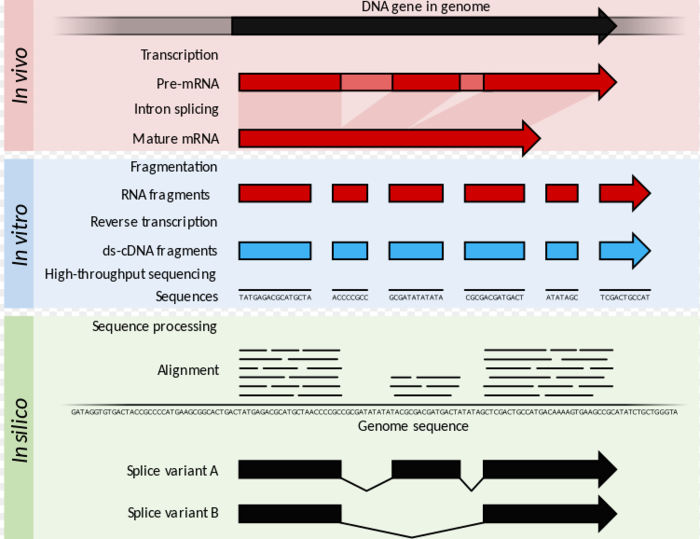
RNA-seq数据分析
mRNA-seq是目前最常用的高通量测序技术,一般的用法就是看看基因表达谱,寻找差异表达的基因。我和高通量测序数据分析结缘,也是因为RNA-seq。
一开始我对mRNA-seq数据分析一无所知,跑了"tophat+cufflinks"的流程也不知道每一步的原因,把“RNA-seq data analysis:A pratice approach” 看了好几遍,也是云里雾里,当然这些时间并没有白白浪费,终于有一天我恍然大悟,感觉自己终于懂了mRNA-seq数据分析,于是在暑假通过一次实战对自己的所学做了一个总结。
- 转录组入门(1):软件准备
- 转录组入门(2):读文章拿到测序数据
- 转录组入门(3):质量控制
- 转录组入门(4):了解参考基因组及基因注释
- 转录组入门(5): 序列比对
- 转录组入门(6): reads计数
- 转录组入门(7):差异表达分析
- 转录组入门(8): 富集分析
但是到目前为止,我实际遇到mRNA-seq数据分析分析项目就一个,不过问我问题的人还是有的,于是打算一边整理实验的流程,再稍微整理下自己的对这方面的理解。
先来看一道RNA-seq数据分析的题目吧,能解决这道题目意味着你真的理解了RNA-seq数据分析。这道问题很简单,不需要强大的计算能力,只需要一张纸和一支笔而已。
这道题目出自 The biostar handbook
假设有一个物种非常的小,仅仅只有三个基因: A, B, C,并且这三个基因都转录本长度分别为10bp, 100bp, 1000bp. 你想通过两个不同的条件下研究该物种,分别是野生型(WT)和热激后(HWEAT)。
由于神秘力量,你知道在WT条件下,基因A的表达量是基因B的表达量的两倍,你还知道在WT和HEAT两个条件中只有一个基因发生了变化(其他基因不变),并且该变化能用目前研究手段中检测到。
你为了找那个在WT和HEAT里不同的基因,非常激动的去做了一次没有重复的RNA-seq实验。由于你很激动,所以不小心把样本混在了一起,而且混了比HEAT处理多一倍WT的DNA量。不过好消息是样本还是能够分开的,毕竟加了barcode。最终结果就是你测了2倍的WT DNA和一倍的HEAT。
问题:你需要准确的用read覆盖情况来表征根据上述给的条件。数字不重要,你可以随便写,重点是这些数字能够表征基因的表达情况。请用实际的数字来替代下面的问号部分
| ID | WT | HEAT |
|---|---|---|
| A | ? | ? |
| B | ? | ? |
| C | ? | ? |
思考题:当你觉得你选择的数字能够回答上面的问题,那么再来想想下面的题目,如果你能回答所有问题,那么那就理解RNA-seq是如何工作的啦。
- 由于你在仪器里放了两倍WT材料,你是如何区分出你的样本?
- 每个条件下,每个基因的CPM是多少?
- 每个条件下,每个基因的RPKM是多少?
- 每个条件下,每个基因的TPM是多少?
- 你怎么知道基因在WT样本中,基因A的表达量真的是基因B表达量的两倍?
- 你能知道WT和HEAT处理中表达量发生变化的基因嘛?
- 当前面的3X2的位置的“?”都有了正确的值,这个问题也是可解决的嘛?
然后,你可以再想想:
- 你需要测多少的read,才能让CPM有一个不错的数值?
- 你需要测多少的read,才能让RPKM有一个不错的数值?
- 你需要测多少的read,才能让TPM有一个不错的数值?
- 你觉得引入上述这些具有任意比例因子的措施是否有意义,还是只为了让数字看起来“很好”?
1F
很详细,赞一个!!
B1
@ PLoB-CAULDD 答案呢 ??
2F
谢谢分享,很有用
3F
RNA-seq数据综合分析教程 有用,学习了
4F
请问有答案吗?求!