

比对软件很多,首先大家去收集一下,因为我们是带大家入门,请统一用hisat2,并且搞懂它的用法。
直接去hisat2的主页下载index文件即可,然后把fastq格式的reads比对上去得到sam文件。
接着用samtools把它转为bam文件,并且排序(注意N和P两种排序区别)索引好,载入IGV,再截图几个基因看看。
前面四篇基本都算是准备工作,从这一篇开始才算进入了RNA-Seq数据分析的核心部分。
比对
比对还是不比对
在比对之前,我们得了解比对的目的是什么?RNA-Seq数据比对和DNA-Seq数据比对有什么差异?
RNA-Seq数据分析分为很多种,比如说找差异表达基因或寻找新的可变剪切。如果找差异表达基因单纯只需要确定不同的read计数就行的话,我们可以用bowtie, bwa这类比对工具,或者是salmon这类align-free工具,并且后者的速度更快。
但是如果你需要找到新的isoform,或者RNA的可变剪切,看看外显子使用差异的话,你就需要TopHat, HISAT2或者是STAR这类工具用于找到剪切位点。因为RNA-Seq不同于DNA-Seq,DNA在转录成mRNA的时候会把内含子部分去掉。所以mRNA反转的cDNA如果比对不到参考序列,会被分开,重新比对一次,判断中间是否有内含子。
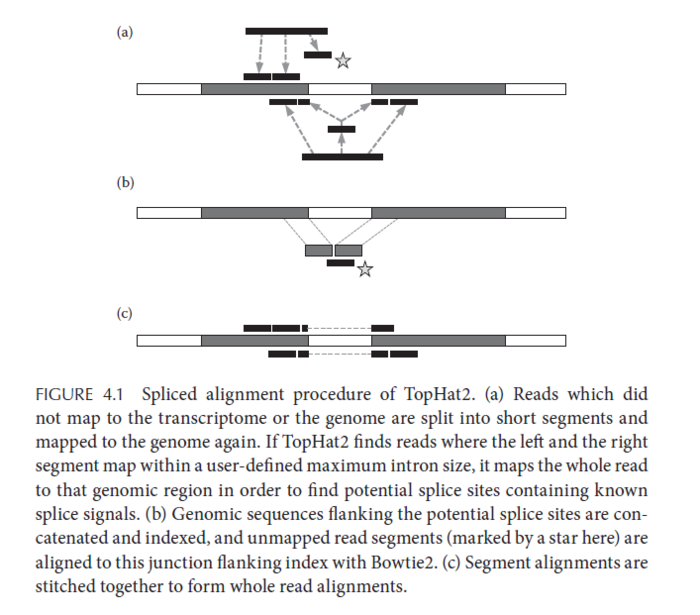
工具抉择
在2016年的一篇综述A survey of best practices for RNA-seq data analysis,提到目前有三种RNA数据分析的策略。那个时候的工具也主要用的是TopHat,STAR和Bowtie.其中TopHat目前已经被它的作者推荐改用HISAT进行替代。
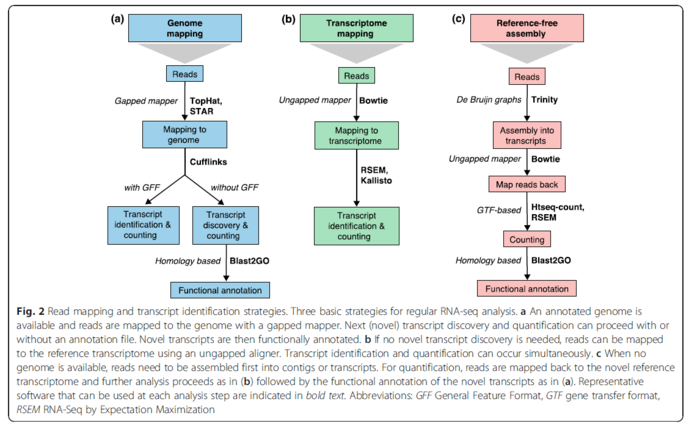
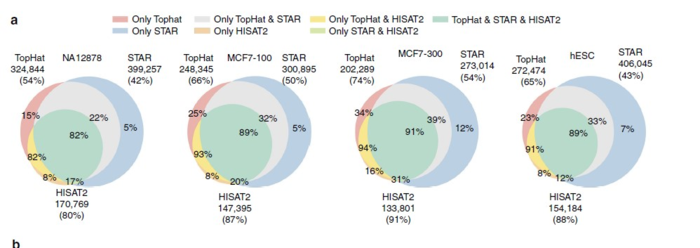
最近的Nature Communication发表了一篇题为的Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis的文章--被称之为史上最全RNA-Seq数据分析流程,也是我一直以来想做的事情,只不过他们做的超乎我的想象。文章中在基于参考基因组的转录本分析中所用的工具,是TopHat,HISAT2和STAR,结论就是HISAT2找到junction正确率最高,但是在总数上却比TopHat和STAR少。从这里可以看出HISAT2的二类错误(纳伪)比较少,但是一类错误(弃真)就高起来。
就唯一比对而言,STAR是三者最佳的,主要是因为它不会像TopHat和HISAT2一样在PE比对不上的情况还强行把SE也比对到基因组上。而且在处理较长的read和较短read的不同情况,STAR的稳定性也是最佳的。
就速度而言,HISAT2比STAR和TopHat2平均快上2.5~100倍。
如果学习RNA-Seq数据分析,上面提到的两篇文献是必须要看上3遍以上的,而且建议每隔一段时间回顾一下。但是如果就比对工具而言,基本上就是HISAT2和STAR选一个就行。
下载index
高通量测序遇到的第一个问题就是,成千上万甚至上几亿条read如果在合理的时间内比对到参考基因组上,并且保证错误率在接受范围内。为了提高比对速度,就需要根据参考基因组序列,经过BWT算法转换成index,而我们比对的序列其实是index的一个子集。当然转录组比对还要考虑到可变剪切的情况,所以更加复杂。
因此我门不是直接把read回贴到基因组上,而是把read和index进行比较。人类的index一般都是有现成的,我建议大家下载现成的,我曾经尝试过用服务器自己创建index,花的时间让我怀疑人生。
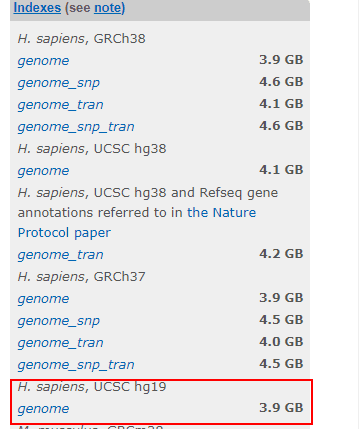
- cd referece && mkdir index && cd index
- wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/data/hg19.tar.gz
- tar -zxvf hg19.tar.gz
觉得电脑配置还行的,或者是没有现成index的,可以通过HISAT2的方法进行创建
- # 其实hisat2-buld在运行的时候也会自己寻找exons和splice_sites,但是先做的目的是为了提高运行效率
- extract_exons.py gencode.v26lift37.annotation.sorted.gtf > hg19.exons.gtf &
- extract_splice_sites.py gencode.v26lift37.annotation.gtf > hg19.splice_sites.gtf &
- # 建立index, 必须选项是基因组所在文件路径和输出的前缀
- hisat2-build --ss hg19.splice_sites.gtf --exon hg19.exons.gtf genome/hg19/hg19.fa hg19
我的是i7-7700处理器,内存是64G,运行的资源效率如下:

正式比对
hisat2基本用法就是hisat2 [options]* -x <hisat2-idx> {-1 <m1> -2 <m2> | -U <r> } [-S <hit>],基本就是提供index的位置,PE数据或者是SE数据存放位置。然而其他可选参数却是进阶的一大名堂。新手就用默认参数呗。
因为RNA-Seq数据是从 SRR3589957 ~ SRR3589962,6个样本的PE数据,也就是有6次循环,可以写脚本,也可以直接在命令行里运行
如下命令运行所在目录为/mnt/f/Data/,我的参考基因组的index数据存放在/mnt/f/Data/reference/index/hg19/,而RNA-seq数据存放在/mnt/f/Data/RNA-Seq下。比对结果会存放在/mnt/f/Data/RNA-Seq/aligned
- mkdir -p RNA-Seq/aligned
- for i in `seq 57 62`
- do
- hisat2 -t -x reference/index/hg19/genome -1 RNA-Seq/SRR35899${i}_1.fastq.gz -2 SRR35899${i}_2.fastq.gz -S RNA-Seq/aligned/SRR35899${i}.sam &
- done
&会把任务丢到后台,所以会同时执行这3个比对程序,如果CPU和内存承受不住,去掉&一个个来。比对这一步是非常消耗内存资源的,这是比对工具要将索引数据放入内存引起的。我有64G内存,理论上可以同时处理20个PE数据。在我的电脑配置下,大致花了2个小时同时才完成这一步.
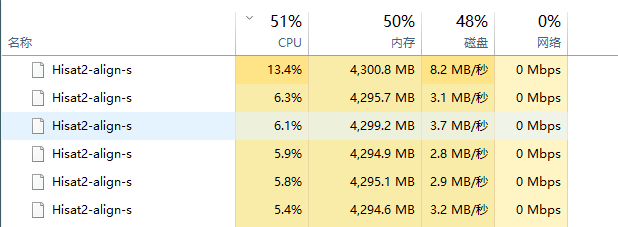
基本参数说明
在数据比对的时候,可以安静一下读读HISAT2的额外选项,主要分为如下几块
- 主要参数,一定要填写的内容
- 输入选项, 对结果影响不大
- 比对选项,主要是
--n-ceil决定模糊字符的数量 - 得分选项, 当一个read比对到不同部位时,确定那个才是最优的。基于mismatch, soft-cliping, gap得分。
- 可变剪切比对选项, 你要决定exon,intron的长度,GT/AG的得分,还可以提供已知的可变剪切和外显子gtf文件,
- 报告选项,确定要找多少的位置
- PE选项, 与gap有关的参数
- 输出选项,建议加上-t记录时间,其他就是压缩格式,不影响比对
- SAM选项, 主要是决定SAM的header应该添加哪些内容
- 性能选项和其他选项不考虑
注: soft clipping 指的是比对的read只有部分匹配到参考序列上,还有部分没有匹配上。也就是一个100bp的read,就匹配上前面20 bp或者是后面20bp,或者是后面20bp比对的效果不太好。
因此影响比对结果就是比对选项,得分选项,可变剪切选项和PE选项,在有生之年我应该会写一片文章介绍这些选项对结果的影响。
HISAT2输出结果
比对之后会输出如下结果,解读一下就是全部数据都是100%的,96.68%的配对数据一次都没有比对,1.23%的数据比是唯一比对,2.09%是多个比对。然后96.68%一次都没有比对的数据,如果不按照顺序来,有0.05%的比对。之后把剩下的部分用单端数据进行比对的话,95.20%数据没比对上,3.60%的数据比对一次,1.20%比对超过一次。零零总总的加起来是8%的比对!!!
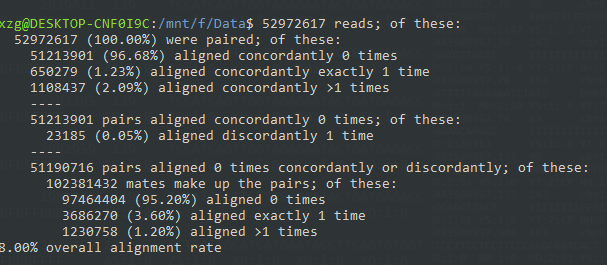
这个总体比对率让我开始怀疑人生,怎么可能呀,我翻了翻输出记录,发现有几个结果的比对率超过90%呀。我思索了片刻,惊醒这个实验好像是用人类和小鼠都做了一遍。于是又去GEO上查了一下记录,恍然大悟,差点翻车。
Samples 9-15 are mRNA-seq to determine effect of AKAP95 knockdown in human 293 cells (9-11) or mouse ES cells (12-15).
同时我反思了一下出错的原因,我默认这个实验是KO和非KO各3个重复,其实文章的实验设计并不是如此,可见理解实验设计很重要,于是我把数据下载这一部分进行了完善。
- mkdir -p RNA-Seq/aligned
- for i in `seq 56 58`
- do
- hisat2 -t -x reference/index/hg19/genome -1 RNA-Seq/SRR35899${i}_1.fastq.gz -2 SRR35899${i}_2.fastq.gz -S RNA-Seq/SRR35899${i}.sam &
- done
如上是修改后的代码
SAMtools三板斧
SAM(sequence Alignment/mapping)数据格式是目前高通量测序中存放比对数据的标准格式,当然他可以用于存放未比对的数据。所以,SAM的格式说明
而目前处理SAM格式的工具主要是SAMTools,这是Heng Li大神写的.除了C语言版本,还有Java的Picard,Python的Pysam,Common lisp的cl-sam等其他版本。SAMTools的主要功能如下:
- view: BAM-SAM/SAM-BAM 转换和提取部分比对
- sort: 比对排序
- merge: 聚合多个排序比对
- index: 索引排序比对
- faidx: 建立FASTA索引,提取部分序列
- tview: 文本格式查看序列
- pileup: 产生基于位置的结果和 consensus/indel calling
最常用的三板斧就是格式转换,排序,索引。而进阶教程就是看文档提高。
- for i in `seq 56 58`
- do
- samtools view -S SRR35899${i}.sam -b > SRR35899${i}.bam
- samtools sort SRR35899${i}.bam -o SRR35899${i}_sorted.bam
- samtools index SRR35899${i}_sorted.bam
- done
注
- -S是最新版samtools为了兼容以前版本写的,所以可以省去
- 0.1.19版本和最新版有比较大差别,请注意版本
我们仔细判断sam排序两种方式的不同,因此我截取前面100行数据,分别排序然后查看结果。
- head -1000 SRR3589957.sam > test.sam
- samtools view -b test.sam > test.bam
- samtools view test.bam | head
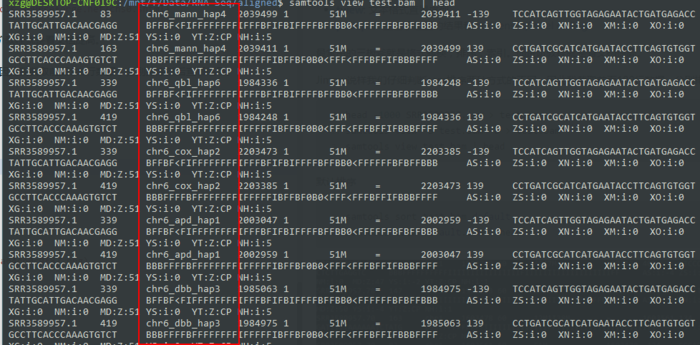
默认排序是根据染色体位置
- samtools sort test.bam default
- samtools view default.bam | head
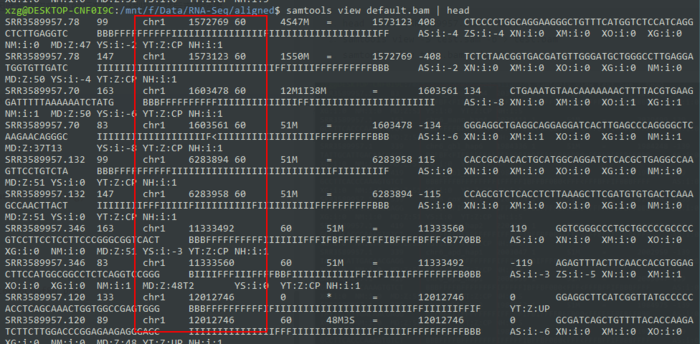
Sort alignments by leftmost coordinates, or by read name when -n is used
- samtools sort -n test.bam sort_left
- samtools view sort_left.bam | head
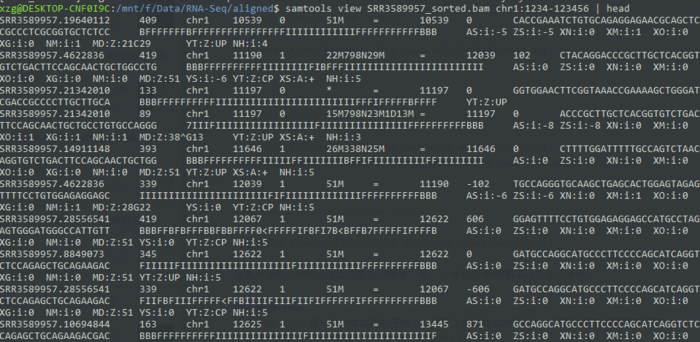
也就说说默认按照染色体位置进行排序,而-n参数则是根据read名进行排序。当然还有一个-t 根据TAG进行排序。
说说samtools view
三板斧的view是一个非常实用的子命令,除了之前的格式转换以外,还能进行数据提取和提取。
比如说提取1号染色体1234-123456区域的比对read
- samtools view SRR3589957_sorted.bam chr1:1234-123456 | head

在比如搭配flag(0.1.19版本没有)和flagstat,使用-f或-F参数提取不同匹配情况的read。
flag是一种描述read比对情况的标记,一种12种,可以搭配使用。
- 0x1 PAIRED paired-end (or multiple-segment) sequencing technology
- 0x2 PROPER_PAIR each segment properly aligned according to the aligner
- 0x4 UNMAP segment unmapped
- 0x8 MUNMAP next segment in the template unmapped
- 0x10 REVERSE SEQ is reverse complemented
- 0x20 MREVERSE SEQ of the next segment in the template is reverse complemented
- 0x40 READ1 the first segment in the template
- 0x80 READ2 the last segment in the template
- 0x100 SECONDARY secondary alignment
- 0x200 QCFAIL not passing quality controls
- 0x400 DUP PCR or optical duplicate
- 0x800 SUPPLEMENTARY supplementary alignment
可以先用flagstat看下总体情况
- samtools flagstat SRR3589957_sorted.bam
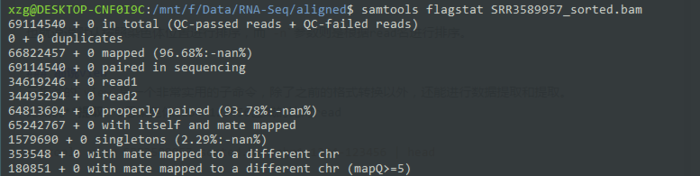
也就是说如果我想用samtools筛选恰好配对的read,就需要用0x10
- samtools view -b -f 0x10 SRR3589957_sorted.bam chr1:1234-123456 > flag.bam
- samtools flagstat flag.bam
我应该会在有生之年写一篇文章好好介绍samtools。
比对质控(QC)
还是在A survey of best practices for RNA-seq data analysis里面,提到了人类基因组应该有70%~90%的比对率,并且多比对read(multi-mapping reads)数量要少。另外比对在外显子和所比对链(uniformity of read coverage on exons and the mapped strand)的覆盖度要保持一致。
常用工具有
- Picard https://broadinstitute.github.io/picard/
- RSeQC http://rseqc.sourceforge.net/
- Qualimap http://qualimap.bioinfo.cipf.es/
我们就用RSeQC吧,毕竟使用python写的工具,天生的亲切感,而且安装非常方便。
- # Python2.7环境下
- pip install RSeQC
一共有如下几个文件,根据命名就知道功能是啥了。
- bam2fq.py
- bam2wig.py
- bam_stat.py
- clipping_profile.py
- deletion_profile.py
- divide_bam.py
- FPKM_count.py
- geneBody_coverage.py
- geneBody_coverage2.py
- infer_experiment.py
- inner_distance.py
- insertion_profile.py
- junction_annotation.py
- junction_saturation.py
- mismatch_profile.py
- normalize_bigwig.py
- overlay_bigwig.py
- read_distribution.py
- read_duplication.py
- read_GC.py
- read_hexamer.py
- read_NVC.py
- read_quality.py
- RNA_fragment_size.py
- RPKM_count.py
- RPKM_saturation.py
- spilt_bam.py
- split_paired_bam.py
- tin.py
先对bam文件进行统计分析, 从结果上看是符合70~90的比对率要求。
- bam_stat.py -i SRR3589956_sorted.bam

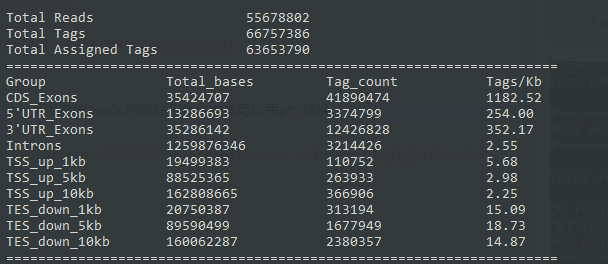
基因组覆盖率的QC需要提供bed文件,可以直接RSeQC的网站下载,或者可以用gtf转换
- read_distribution.py -i RNA-Seq/aligned/SRR3589956_sorted.bam -r reference/hg19_RefSeq.bed
IGV查看
载入参考序列,注释和BAM文件,随便看看吧。
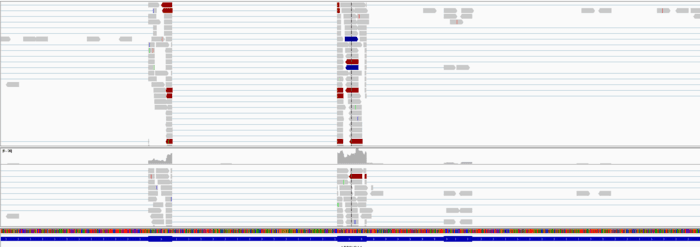
1F
转录组入门(5): 序列比对