

VCF文件格式
在进行变异检测后我们最后以VCF格式存放找到的变异。尽管大部分情况下,我们都不需要直接和VCF文件打交道,通常就是将其作为输入提供给后续的分析。但是,你对VCF的格式越熟悉,你就能使用bcftools,vcftools或其他工具提取你任意需要的数据。
VCF(Variant Call Format)可以用来存放找到的变异信息,包括三个部分,元信息,标题行和数据行。
举个例子
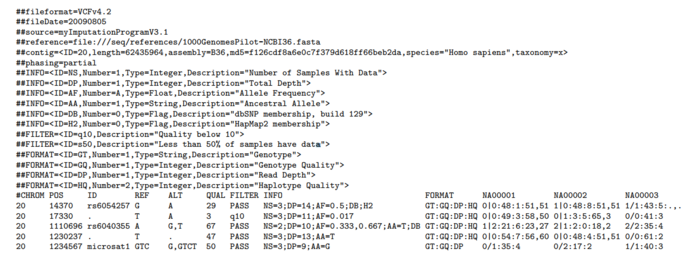
元信息(meta-information)行以"##"起始,首先是VCF的版本信息,后面的INFO定义和解释INFO列出现的ID的含义,FILTER解释说明做了过滤的类型,而FORMAT则解释FORMAT列出现的ID的含义和数据结构,SAMPLE则是告知有哪些样本,都比较的浅显易懂。
标题列固定8列,CHROM(染色体ID), POS(变异所在位置), ID(已有注释), REF(参考碱基), ALT(变异碱基), QUAL(质量),FILTER(过滤方式),INFO(总体信息列),第9列为FORMAT,定义了后面每个样本的数据存放结构。
对于数据行,我比较在意的是如何在VCF存放和解读变异信息,所以也重点介绍这部分。
对于SNP和较小的indels
对于比较小的SNP,或者是插入缺失的碱基在20bp以内的indel,表示比较容易,读起来也不太费劲。
比如说在参考基因组和样本的基因组上某个位置上有如下情况
| 案例 | 序列 | 说明 |
|---|---|---|
| Ref | a t C g a | 参考序列 |
| 1 | a t G g a | C突变成G |
| 2 | a t - g a | C缺失 |
| 3 | a t CAg a | C后插入A |
如果只有一个样本,表示方法位:
- #CHROM POS ID REF ALT QUAL FILTER INFO # 解读
- 20 3 . C G . PASS DP=100 # ref第三位是C,而ALT第三位是G
- 20 2 . TC T . PASS DP=100 # ref第二位开始时TC,而ALT第二位开始只有T,说明ALT的第三位C缺失
- 20 2 . TC TCA . PASS DP=100 # ref第二位是TC,而ALT第二位开始时TCA,说明多了一个A
如果同时表示三个样本
- #CHROM POS ID REF ALT QUAL FILTER INFO # 解读
- 20 2 . TC TG,T,TCA . PASS DP=100 # 表示在该位点上有三个突变信息
那么已知ref为atCga,根据VCF信息进行重组
- #CHROM POS ID REF ALT QUAL FILTER INFO # 重组结果
- 20 3 . C T . PASS DP=100 # atTga
- 20 3 . C CTAG . PASS DP=100 # atCTAGga
- 20 2 . TCG T . PASS DP=100 # aTa
结构变异(structure variants)
这里的定义SV,指的是插入缺失大于20bp,小于12kb的情况,先随意感受下VCF是如何处理这种情况。
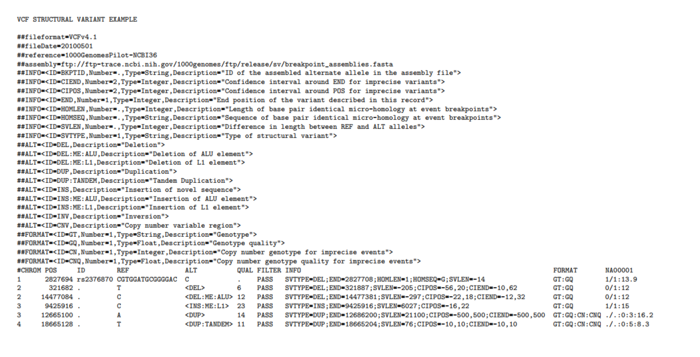
即为了表示SV,需要专门定义INFO和FORMAT。根据定义就能对这6个变异进行解读
- 一个准确的缺失, 发生在2827694-2827708
- 一个不太准确的缺失(DEL),长度大概在205bp(SVLEN=-205)
- 一个不太准确的ALU缺失(DEL:ME:ALU),长度大概为209bp(SVLEN=-209)
- 一个不太准确的LA插入(INS:ME:L1)
- 也差不多
大规模重排
暂时不在讨论范围之内。
BCFtools
BCFtools是一套处理VCF和BCF格式的工具。它有提供许多子命令实现不同功能,我个人用的比较多有以下几个:
- mpileup + call: 根据参考基因组寻找变异位点
- view: 选取,过滤以及VCF/BCF之间的格式转换。 这个命令完成了
convert,filter, - query: VCF/BCF格式输出为更适合人类阅读的格式
- merge: 将多个VCF/BCF文件整合成一个
- isec: 求不同VCF/BCF文件的交集,合集和补集
通用参数
在单独介绍每个命令之前,需要了解一下所有子命令都可以用的参数:
文件输出: -o, --ouput FILE,默认输出到标准输出,通过该选项指定文件。 -O, --output-type b|u|z|v: 输出为压缩的BCF(b), 未压缩的BCF(u), 压缩的VCF(z), 未压缩的VCF(v). 使用-Ou能够让bcftools命令间的操作更快。
- FILE:输入文件,可以是VCF或BCF,以及这些文件对应的BGZIP压缩形式。如果是
-, 则认为是标准输入。有些工具需要tabix或CSI的索引文件。
mpileup和call是一套组合,最基本的用法为:
- bcftools mpileup -Ou -f reference.fa alignments.bam | bcftools call -mv -Ob -o calls.bcf
- # -m: 允许多倍体
- # -v:表示只输出和基因组不同的位点
根据不同情况可以添加call的参数,比如说 bcftools call -P 1.1e-3。
view一般要配合-f LIST参数共同使用,能有效对VCF/BCF文件内容进行筛选。比如说仅选择非indel, 且ref的reads数小于1, 深度在20和100之间。
- bcftools view -i 'TYPE!="indel" && (DP4[0]+DP4[1])<1 && DP >20 && DP < 100'
有效表达式包括:
- 数值常量,字符常量和文件名:
1,1.0,1e-4,"string",@filename - 算术运算符:
+, *, -, / - 比较操作符:
==, >, >=, <, <=, != - 正则表达式:
INFO/HAYSTACK ~"needle/i" - 括号:
() - 逻辑运算符:
&&, || - INFO标签和FORMAT标签以及列名
- INFO/DP 或 DP
- FORMAT/DV, FMT/DV, 或 DV
- FILTER, QUAL, ID, POS, REF, ALT[0]
- 1或0用于判断flag是否存在
- "."则是判断是否有缺失值
- 样本基因型: 纯合("hom"),杂合("het"),单倍体("hap"),alt-alt纯合("AA"),ref-alt杂合("RA"),alt-alt杂合("Aa"),单倍体参考("R"),单倍体替换("A")
- REF/ALT列的变异类型(TYPE): indel, snp, mnp, ref, bnd, other
- 数组下标: (DP[0]+DP[1])/(DP[2]+DP[3]) > 0.3。 其中
*表示任意,-表示返回 - FORMAT和INFO标签的函数: MAX, MIN, AVG, SUM, STRLEN, ABS
- 运行过程中新增变量:
N_ALT, N_SAMPLES, AC, MAC, AF, MAF, AN, N_MISSING, F_MISSING
query可以将VCF/BCF文件转换成更加人类可读的格式,依赖于-f FORMAT参数。
其中FORMAT可以是:
- 所有的列名:%CHROM, %POS, %ID, %REF, %ALT, %QUAL, %FILTER
- INFO列的其中一个:%INFO/标签(如INFO/DP4),此外标签是多值结果,能用
{}进一步选取,例如DP4{1} - 基因型: "%GT", "%TGT"
- 换行符和制表符:"\n","\t"
实际操作
后续操作需要下载案例数据
- curl -O http://data.biostarhandbook.com/variant/subset_hg19.vcf.gz
- curl -O http://data.biostarhandbook.com/variant/subset_hg19.vcf.gz.tbi
从VCF中按照自定义格式提取数据
- bcftools query -f '%CHROM %POS %REF %ALT \n' subset_hg19.vcf.gz | head -3
- # 结果
- 19 400410 CA C
- 19 400666 G C
- 19 400742 C T
列出存放的所有样本
- bcftools query -f subset_hg19.vcf.gz
从指定区域提取所有变异位点
- bcftools query -f '19:400300-400800' -f '%CHROM\t%POS\t%REF%ALT\n' subset_hg19.vcf.gz | head -3
- 19 400410 CAC
- 19 400666 GC
- 19 400742 CT
这里是按照特定格式提取,如果希望输出也是VCF文件,则用filter或view命令。
从指定区域外提取所有变异
- bcftools view -H -t ^'19:400300-400800' subset_hg19.vcf.gz | head -3
- # 结果如下
- 19 400819 rs71335241 C G 100 PASS AC=0;AF=0.225839;AN=12;NS=2504;DP=10365;EAS_AF=0.2897;AMR_AF=0.2349;AFR_AF=0.2088;EUR_AF=0.161;SAS_AF=0.2434;AA=N|||;VT=SNP GT 0|0 0|0 0|0 0|0 0|0 0|0
- 19 400908 rs183189417 G T 100 PASS AC=1;AF=0.0632987;AN=12;NS=2504;DP=13162;EAS_AF=0.002;AMR_AF=0.1153;AFR_AF=0.0726;EUR_AF=0.0885;SAS_AF=0.0511;AA=-|||;VT=SNP GT 0|0 0|0 0|0 0|0 0|0 0|1
- 19 400926 rs28420134 C T 100 PASS AC=1;AF=0.0259585;AN=12;NS=2504;DP=13731;EAS_AF=0.005;AMR_AF=0.0879;AFR_AF=0.003;EUR_AF=0.0457;SAS_AF=0.0143;AA=C|||;VT=SNP GT 0|0 0|0 0|0 0|0 0|1 0|0
根据样本中的基因型信息提取
- # 通过表达式
- bcftools view -e 'GT="." | GT="0|0"' subset_hg19.vcf.gz | bcftools query -f '%POS[\t%GT\t]\n' | head -3
- 402556 0|1 0|1 1|1 1|0 0|1 1|1
- 402707 0|1 0|1 1|1 1|0 0|1 1|1
- 402723 0|1 0|1 1|1 1|0 0|1 1|1
- # 或者是-g/--genotype
- ## 选择至少有一个样本是杂合,且所有样本都不包含缺失位点信息
- bcftools view -g het subset_hg19.vcf.gz | bcftools view -g ^miss | bcftools query -f '%POS[\t%GT]\n' | head -3
仅提取INDEL, 可用-v/--type或-i/--include, 当然这两者有细微区别。
- bcftools view -v indels subset_hg19.vcf.gz | bcftools query -f '%POS\t%TYPE\n' | wc -l
- bcftools view -i 'TYPE="indel"' subset_hg19.vcf.gz | bcftools query -f '%POS\t%TYPE\n' | wc -l
仅选择或不选择某几个样本
- bcftools view -s HG00115,HG00118 subset_hg19.vcf.gz | bcftools query -H -f '%POS[\t%GT]\n' | head -n 4
- bcftools view -s ^HG00115,HG00118 subset_hg19.vcf.gz | bcftools query -H -f '%POS[\t%GT]\n' | head -n 4
选择等位基因大于或者低于一定值的变异,即比较AC(alternate alleles count)
- # 大于5
- bcftools view -c 5 subset_hg19.vcf.gz | bcftools query -f '%POS[\t%GT]\n' | head
- ## 结果如下
- 400666 1|0 0|1 0|1 0|0 0|0 1|1
- 401818 0|1 0|1 1|1 1|0 0|0 1|1
- 401907 0|1 0|1 1|0 1|0 0|0 0|1
- # 低于5
- bcftools view -C 5 subset_hg19.vcf.gz | bcftools query -f '%POS[\t%GT]\n' | head -3
- ## 结果如下
- 400410 0|0 0|0 0|0 0|0 0|0 0|0
- 400666 1|0 0|1 0|1 0|0 0|0 1|1
- 400742 0|0 0|0 0|0 0|0 0|0 0|0
根据变异质量和覆盖深度选择
- bcftools query -i 'QUAL>50 && DP>5000' -f '%POS\t%QUAL\t%DP\n' subset_hg19.vcf.gz | head -3
- 400410 100 7773
- 400666 100 8445
- 400742 100 15699
对于多个VCF文件,则需要用到merge和isec。
- # 合并列表中的样本
- bcftools merge -l samplelist > multi-sample.vcf
- # 提取在所有样本都出现的变异
- bcftools isec -p outdir -n=3 sample1.vcf.gz sample2.vcf.gz sample3.vcf.gz
- # 提取至少在两个样本出现的变异
- bcftools isec -p outdir -n+2 sample1.vcf.gz sample2.vcf.gz sample3.vcf.gz
- # 提取仅仅在一个样本中出现的变异
- bcftools isec -p outdir -C sample1.vcf.gz sample2.vcf.gz sample3.vcf.gz
输出结果就能直接导入到R,Python进行分析。
VCFtools
VCFtools: 用于描述性统计数据,计算数据,过滤数据以及数据格式转换。
基本用法:
- vcftools [--vcf VCF文件 | --gzvcf gz压缩的VCF文件 --bcf BCF文件] [--out OUTPUT PREFFI]
他能做的事情:
- 输出第一条染色体的所有位点等位基因频率
- 从输入文件中仅保留SNP位点
- 输出两个vcf文件的比较结果
- 标准输出不含有filer tag的位点,并且以gzip压缩
- 计算每个位点的hardy-weinberg p-value,这些位点不包括缺失的基因型
- 计算一系列核酸多态性
常用参数如下:
- 和输入输出有关
- --vcf, --gzvcf, --bcf:根据输入文件格式进行选择
- --out, --stdout, -c --temp: 选择合适的输出方式
- 位点筛选(site filtering)有关参数
- # 根据位置过滤
- --chr/--not-chr Chr1: 选择染色体
- --from-bp/--to-bp: 选择碱基范围
- # 根据第三列ID进行过滤,不常用
- --snp rsID
- # 根据变异类型
- --keep-only-indels: 仅保留INDEL
- --remove-indels: 仅保留SNP
- # 根据FILTER列进行过滤
- --remove-filtered-all: 移除filter tag位点
- --keep-filtered/--remove-filtered
- # 根据INFO列进行过滤
- --keep-INFO/--remove-INFO 目标类型
- # 根据等位基因过滤
- --maf/--max-maf # Minor Allele Frequency
- --mac/--max-mac # Minor Allele Count
- 样本样本参数(我不常用)
- 基因型过滤参数(没怎么用)
- 输出VCF选项
- --recode
- --recode-INFO-all
- --diff-site: 比较位点
- --hardy: hardy-weinberg p值
- --max-missing: 基因型缺失
- --site-pi