

本系列教程包括:
基因表达
什么是基因表达,如下是来自于维基百科的解释:
Gene expression is the process by which information from a gene is used in the synthesis of a functional gene product. These products are often proteins, but in non-protein coding genes such as transfer RNA (tRNA) or small nuclear RNA (snRNA) genes, the product is a functional RNA. The process of gene expression is used by all known life—eukaryotes (including multicellular organisms), prokaryotes (bacteria and archaea), and utilized by viruses—to generate the macromolecular machinery for life.

研究方法
定量PCR
这部分我不太懂,所以就放几段百度百科和维基百科的定义。
- 百度百科
定量PCR(即时聚合酶链锁反应,Real-time Polymerase Chain Reaction,简称 Real-time PCR、即时PCR),又称定量即时聚合酶链锁反应(Quantitative real time polymerase chain reaction,简称 Q-PCR/qPCR/rt-qPCR、定量即时PCR、即时定量PCR),是一种在DNA扩增反应中,以萤光染剂侦测每次聚合酶链锁反应(PCR)循环后产物总量的方法技术,有广义概念和狭义概念。广义概念的定量PCR技术是指以外参或内参为标准,通过对PCR终产物的分析或PCR过程的监测,进行PCR起始模板量的定量。狭义概念的定量PCR技术(严格意义的定量PCR技术)是指用外标法(荧光杂交探针保证特异性)通过监测PCR过程(监测扩增效率)达到精确定量起始模板数的目的,同时以内对照有效排除假阴性结果(扩增效率为零)。
- 维基百科
A real-time polymerase chain reaction (Real-Time PCR), also known as quantitative polymerase chain reaction (qPCR), is a laboratory technique of molecular biology based on the polymerase chain reaction (PCR). It monitors the amplification of a targeted DNA molecule during the PCR, i.e. in real-time, and not at its end, as in conventional PCR. Real-time PCR can be used quantitatively (quantitative real-time PCR), and semi-quantitatively, i.e. above/below a certain amount of DNA molecules (semi quantitative real-time PCR).
优点:灵敏性高,准确性高,通量也还行。一般而言,RNA-Seq和microassay分析得到的差异表达基因最终也需要通过这种实验方法进行验证。
但是一般适用于验证实验,而不是用于探索性实验。
microarray<small>基因矩阵</small>
基因芯片的概念在上个世纪80年代就已经提出来了, 被评为1998年度自然科学领域十大进展之一。他的基本原理通过设计专门的短核苷酸作为探针,把这些探针固定在专门的基片表面,然后用样本的cDNA进行杂交,根据杂交信号的强弱来判断基因表达的程序。

但是microarray检测的基因数量完全取决于你的探针设计的数量,而且难以研究mRNA的可变剪切。
RNA-Seq
RNA-Seq是目前基因表达分析最常用的技术。分为以下几步
- 分离所有mRNA
- 逆转录mRNA成cDNA
- 对cDNA测序
- 比对参考基因组
RNA-Seq实验设计中的“重复”包括:技术重复和生物学重复
重复是为了检测组间和组内的变异,对于假设检验至关重要。
- 技术重复为了估计测量技术(RNA-Seq)的变异。
- 生物学重复是为了发现生物组内的变异。
简单的说,两组的基因表达的变化只有比组内变异还大时才能认为时显著的。
RNA-Seq的概率分布
相同基因在不同细胞的表达水平服从log-normal(对数正态)分布,由定量PCR验证。(注:这与相同细胞不同基因表达的分布不同)但是大多数基因表达实验都是用一群细胞,几乎没有相应分布提出。
RNA-Seq试验中,抽样得到的raw read counts服从泊松分布。并且同一样本在两次试验中的结果不同,这称为shot noise。这种变异在RNA-Seq技术重复间成为Possion noise。
生物学上不同的样本间的差异服从负二项(negative binomial)分布,有时称gamma-Poisson分布。
由于RNA-Seq count数据也表现出zero inflation(大量值为0)的特征,所以很难拟合到负二项分布,所以有文章认为要用Poisson-Tweedie family建模。
RNA-seq数据和microassay在差异表达分析上的区别:
- RNA-Seq观察到的数据是抽样过程中产生的离散(discrete)count形式。也就是说总体是恒定的,表达量越高的基因在抽样结果中所占的比例越大。表达量低的基因可能即便有也无法被检测出来。当然,重新对相同文库进行测序,还是有可能找到更多表达的转录本
- microassay检测的是荧光信号的连续度量。由于使用固定的核酸序列去杂交,所以不是一种“零和游戏”,只要能杂交,就能被检测。(但如果没有设计相应的引物,就不能检测到可能的基因)
研究意义
1.在不同背景下比较mRNA水平
- 同一物种,不同组织:研究基因在不同部分的表达情况
- 同一物种,同一组织:研究基因在不同处理下,不同条件下的表达变化
- 同一组织,不同物种:研究基因的进化关系
- 时间序列实验: 基因在不同时期的表达情况与发育的关系
2.基因分类: 找到细胞特异,疾病相关,处理相关的基因表达模式,用于诊断疾病和预测等
3.基因网络和通路: 基因在细胞活动中的功能,基因间的相互作用。
公共数据库
当然,如果你要研究一个基因的功能时,不要先急着去花钱找公司测序,先去一些基因表达公共数据库找找看:
差异表达(differential expression,DE)基因分析
通过研究基因的差异表达,我们可以发现
- 细胞特异性的基因;
- 发育阶段特异性的基因;
- 疾病状态相关的基因;
- 环境相关的基因;
- ...
基本方法就是以生物学意义的方式计算基因表达量,然后通过统计学分析表达量寻找具有统计学显著性差异的基因,从而
- 选择合适的基因
- 衡量结果的可靠性
分析方法
寻找差异表达基因有三种方式:
第一种是计算Fold change(倍数变化),十分简单粗暴的方法,计算方法如下:
- E = mean(group1) B=mean(group2)
- FC = (E-B) / min(E,B)
说人话就是,基因A和基因B的平均值之差与两者中较小的比值。选择2-3倍的基因作为结果(为什么是2-3倍,就是大家约定俗成)。
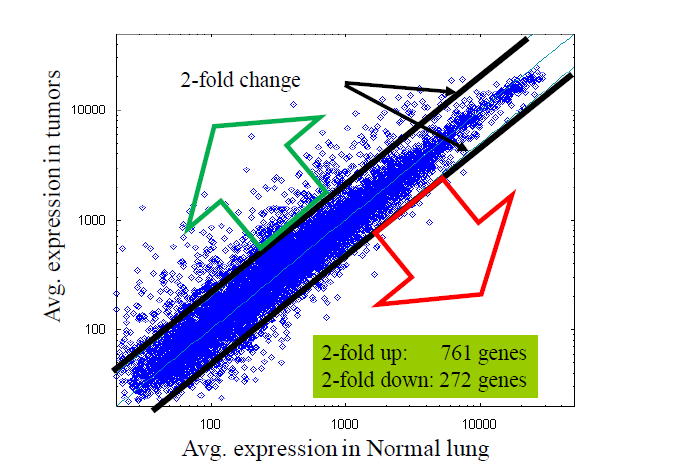
但是简单粗暴的用2到3倍作为阈值,对于低表达的基因,3倍也是噪音,那些高表达的基因,1.1倍都是生物学显著了。更重要的没有考虑到组内变异,没有统计学意义。所以发文章肯定这个图只能作为附录了。
第二种就是统计检验,写文章的时候总需要给出一个p值告诉主编这个结果可信的(虽然p值也存在争论)。
复习一下:p值指的碰巧是拒绝零假设机会。P值越大假阳性越低,同时真实结果也可能会剔除。
注: 基因表达分析的零假设是: 基因在不同处理下的表达量相同。
对于基因芯片的数据而言,由于样本服从正态分布,所以可以用t-test(双处理)或anova分析(多处理以上)。
T检验适用于只有两个处理的实验设计,如植物叶片在相同处理第一天和第二天的基因表达差异。
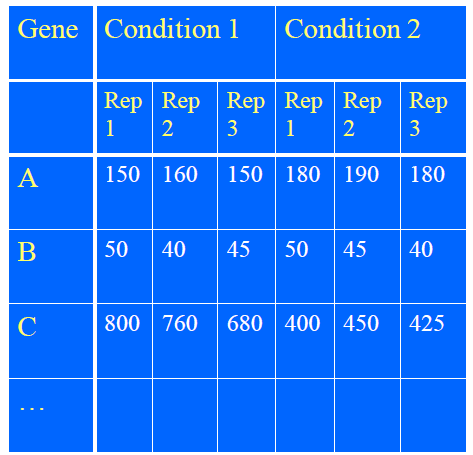
进行T-test检验时要注意:是双尾检验(存在差异)还是单尾检验(显著性上调或下降),两个样本的总体是不是等方差(标准T检验还是Welch’s test)
如果存在多于两个处理(条件),就需要用到ANOVA分析了。ANOVA分析能主要是研究结果之间的差异是如何引起的,具体请移步到我写方差分析教程。
对于基因表达而言,研究目标是,对于同一个基因而言,他们之间的差异是处理不同造成,还是因为系统误差造成。
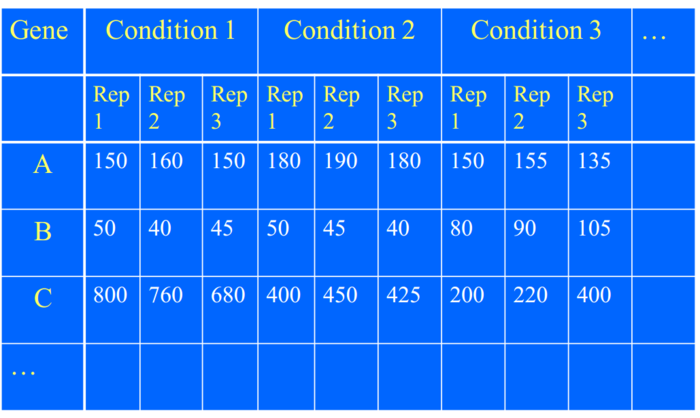
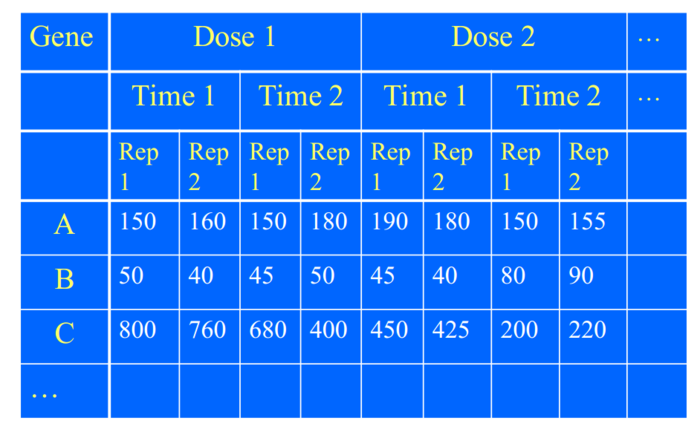
当然你可以研究,不同基因的表达差异是由因为处理不同,还是基因不同,还是系统误差,还是其中一些的交互作用。
上面都是针对基因芯片的样本服从正态分布进行的统计检验。现在的RNA-Seq,它的抽样过程是离散的,结果是count,服从泊松分布,样本间的差异是服从负二向分布,显然不能按照上述方法分析。
方差分析(ANOVA)和线性回归分析(regression)都是同一时期发展的两套紧密相连的理论。方差分析考量的是离散型自变量(因子)对连续型应变量(响应变量)的模型分析,而线性回归分析只要求响应变量是连续的,对于自变量无要求。如果响应变量不是连续型分布,就要使用更加一般化的广义线性模型(generalized linear model),通过一个连接函数变换响应变量期望,将响应变量的期望与自变量建立线性关系。
因此,我们可以用广义线性模型去分析RNA-seq前期分析得到的离散型结果(count)
方差分析一般用于分析有计划的实验结果,比如说不同处理下的水稻产量。回归分析一般分析没有计划的数据,比如说你可以找到大量体检的数据,只分析其中性别和身高对体重的影响。所以两者各有侧重,不要拿大炮轰蚊子。
统计检验相对于fold change具有统计意义,不需要参考样本,需要处理随机取样。但是需要重复(ANOVA推荐4-10重复),但由于资金和材料等原因,不一定能够满足。此外,对于1000个基因,就要做1000次ANOVA或t-test,最后的p值会有一定的假阳性,因此要做p值矫正(FDR)筛选。
注:推荐在统计检验前过滤表达量低,也就如果一个基因在所有样本中count均低于某一阀值,请在分析前剔除。这个阀值也是约定俗成,一般设置为3.
第三种:Fold Change + 统计检验。说一比较尴尬的事情,在统计检验中你找到越多的差异表达基因,在p值矫正之后,你反而找不到差异表达基因。也就是说,如果在结果中存在大量滥竽充数的所谓的DE基因,那么在严格的p值矫正筛选后,反而会误删真实的DE基因。
因此在p值矫正之前,你先要手动剔除一部分明显就是假阳性的DE基因。这个步骤就需要用到前面的fold-change分析。
我们可以通过火山图来看看如何确定区间
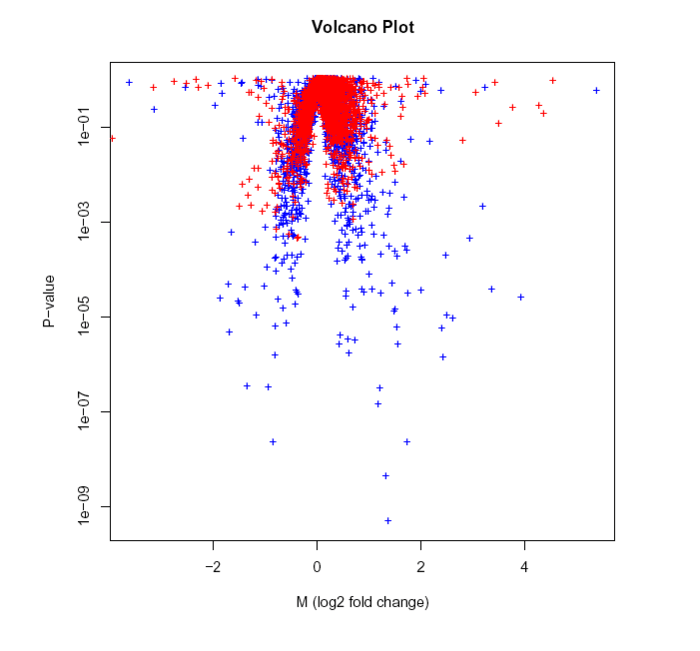
实战演练
为了方便理解,我们选用同一组数据进行实际操作。默认你懂基本的R语言操作,如安装R包,查看帮助文件等。
正式学习之前,先感谢一下Bioconductor。 根据他们提供流程,我学习到如何进行RNA-Seq分析。
- http://www.bioconductor.org/help/workflows/RnaSeqGeneEdgeRQL/
- http://www.bioconductor.org/help/workflows/rnaseqGene/
- http://www.bioconductor.org/help/workflows/RNAseq123/
- http://www.bioconductor.org/help/workflows/ExpressionNormalizationWorkflow/
数据介绍和下载
用到的数据来自于一篇文献“A pooled shRNA screen for regulators of primary mammary stem and progenitor cells identifies roles for Asap1 and Prox1”。
实验目的:
The aim of this study is to determine the absolute and relative expression levels of mRNA transcripts across multiple flow cytometrically sorted epithelial cell types including freshly isolated CD24+CD29hi mammary stem cell-enriched basal cells (MaSC/basal), CD24+CD29loCD61+ luminal progenitor-enriched (LP) and the CD24+CD29loCD61- mature luminal-enriched (ML) cell populations. Additionally, a comparison between these primary cell types and cultured MaSC/Basal-derived mammosphere cells (mammosphere) and the CommaD-βGeo (CommaDβ) cell line was performed.
实验设计:
Total RNA was extracted and purified from sorted luminal or basal populations from the mammary glands of female virgin 8- to 10-week-old FVB/N mice (3 independent samples for population), MaSC/Basal cells cultured for 1 week under mammsophere conditions and CommaDβ cells grown under maintenance conditions (Deugnier et al. 2006) and their transcriptomes analysed by RNA-Seq. |
根据文章的介绍,数据是100 bp的单端read,用Rsubread比对到mouse reference genome(mm10), 然后使用featureCounts统计每个基因的count数。然后用TMM进行标准化,转换成log2 counts per million.最后用limma包对每个样本每个基因的平均表达值以观察水平权重的线性模型进行拟合,并用T检验找到不同群体的差异表达基因。以FDR + log2-fold-change对基因排序。
我们的任务是下载他们featureCounts的结果,不用limma,改用DESeq2分析。
- # install.packages("R.utils")
- library("R.utils")
- url <- "https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE63310&format=file"
- utils::download.file(url, destfile="GSE63310_RAW.tar", mode="wb")
- utils::untar("GSE63310_RAW.tar", exdir = ".")
- files <- c("GSM1545535_10_6_5_11.txt", "GSM1545536_9_6_5_11.txt", "GSM1545538_purep53.txt",
- "GSM1545539_JMS8-2.txt", "GSM1545540_JMS8-3.txt", "GSM1545541_JMS8-4.txt",
- "GSM1545542_JMS8-5.txt", "GSM1545544_JMS9-P7c.txt", "GSM1545545_JMS9-P8c.txt")
- for(i in paste(files, ".gz", sep=""))
- R.utils::gunzip(i, overwrite=TRUE)
数据读取
先定一个包含所有文件的向量,方便后续调用。
- files <- c("GSM1545535_10_6_5_11.txt", "GSM1545536_9_6_5_11.txt",
- "GSM1545538_purep53.txt", "GSM1545539_JMS8-2.txt",
- "GSM1545540_JMS8-3.txt", "GSM1545541_JMS8-4.txt",
- "GSM1545542_JMS8-5.txt", "GSM1545544_JMS9-P7c.txt",
- "GSM1545545_JMS9-P8c.txt")
查看下文件的数据格式,分别记录了每个基因的EntrezID,基因长度和数量
- read.delim(files[1], nrow=5)
- # EntrezID GeneLength Count
- #1 497097 3634 1
- #2 100503874 3259 0
- #3 100038431 1634 0
- #4 19888 9747 0
- #5 20671 3130 1
之后我们需要用DESeqDataSetFromMatrix为DESeq2提供数据,命令形式如下:
- library("DEseq2")
- DESeqDataSetFromMatrix(countData, colData, design, tidy = FALSE,ignoreRank = FALSE, ...)
其中countData存放counts数据,colData存放样本信息的数据,design就是实验设计。
首先从各个count matrix文件中读取count,基因长度部分可以舍弃,因为DESeq2只需要为标准化的count数据,不需要提供基因长度信息。
逻辑就是分别读取每一个文件的count列,然后赋予文件名。
- sample1 <- read.delim(files[1],header = T)[,c(1,3)]
- count.table <- data.frame(sample1)
- for ( f in files[2:length(files)]){
- column <- read.delim(f,header=T,row.names = 1)[,2]
- count.table <- cbind(count.table, column)
- }
- head(count.table)
- rownames(count.table) <- count.table[,1]
- count.table <- count.table[-1]
- samplenames <- substring(files, 12, nchar(files)-4)
- colnames(count.table) <- samplenames
然后要提供colData,其中colData存放列名要和countData的行名相同。
- # colData
- condition <- as.factor(c("LP", "ML", "Basal", "Basal", "ML", "LP",
- "Basal", "ML", "LP"))
- lane <- as.factor(rep(c("L004","L006","L008"), c(3,4,2)))
- colData <- data.frame(condition = condition, lane=lane,row.names = samplenames)
- all(rownames(colData) %in% colnames(count.table))
最后导入数据:
- dds <- DESeqDataSetFromMatrix(countData = count.table,
- colData = colData,
- design = ~ condition )
数据预处理
预过滤
虽然DESeq2会自动屏蔽那些低count的基因,但是剔除那些几乎不存在基因的部分能够提高运行速度。
- dds <- dds [ rowSums(counts(dds)) > 1, ]
rlog和方差齐性转换
许多常见的多维数据探索性分析的统计分析方法,例如聚类和主成分分析要求,在那些同方差性的数据表现良好。所谓的同方差性就是虽然平均值不同,但是方差相同。
但是对于RNA-Seq count数据而言,当均值增加时,方差期望也会提高。也就说直接对count matrix或标准化count(根据测序深度调整)做PCA分析,由于高count在不同样本间的绝对差值大,也就会对结果有很大影响。简单粗暴的方法就是对count matrix取log后加1。这个1也是约定俗成,看经验了。
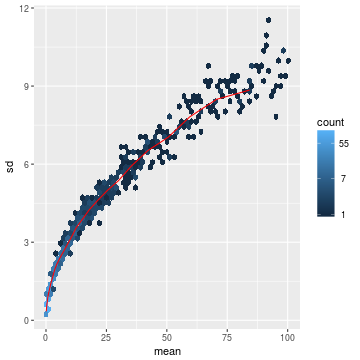
随便举个栗子看下效果:
- lambda <- 10^seq(from = -1, to = 2, length = 1000)
- cts <- matrix(rpois(1000*100, lambda), ncol = 100)
- library("vsn")
- meanSdPlot(cts, ranks = FALSE)
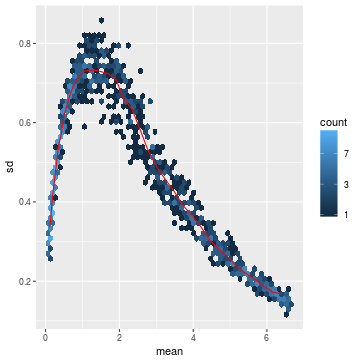
- log.cts.one <- log2(cts + 1)
- meanSdPlot(log.cts.one, ranks = FALSE)
DESeq2为count数据提供了两类变换方法,使得不同均值的方差趋于稳定:regularized-logarithm transformation or rlog(Love, Huber, and Anders 2014)和variance stabilizing transformation(VST)(Anders and Huber 2010)用于处理含有色散平均趋势负二项数据。
到底用啥:数据集小于30 -> rlog,大数据集 -> VST。还有这个处理过程不是用于差异检验的,在DESeq分析中会自动选择最合适的所以你更不需要纠结了,记得用raw count。
- rld <- rlog(dds, blind = FALSE)
- head(assay(rld), 3)
- vsd <- vst(dds, blind = FALSE)
- head(assay(vsd), 3)
处理的结果如下:
- library("dplyr")
- library("ggplot2")
- dds <- estimateSizeFactors(dds)
- df <- bind_rows(
- as_data_frame(log2(counts(dds, normalized=TRUE)[, 1:2]+1)) %>%
- mutate(transformation = "log2(x + 1)"),
- as_data_frame(assay(rld)[, 1:2]) %>% mutate(transformation = "rlog"),
- as_data_frame(assay(vsd)[, 1:2]) %>% mutate(transformation = "vst"))
- colnames(df)[1:2] <- c("x", "y")
- ggplot(df, aes(x = x, y = y)) + geom_hex(bins = 80) +
- coord_fixed() + facet_grid( . ~ transformation)
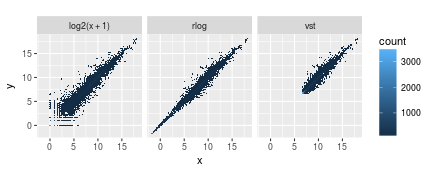
结果就是转换后更加集中了。
样本距离
RNA-Seq分析第一步通常是评估样本间的总体相似度。
- 那些样本最接近
- 那些样本差异较大
- 这与实验设计预期符合么
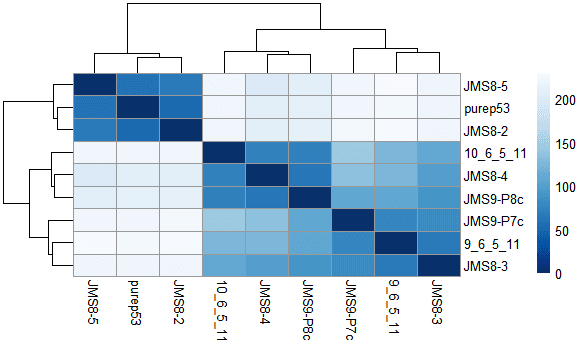
这里使用R内置的 dist 计算 rlog变换数据的Euclidean distance,然后用pheatmap根据结果画热图
- sampleDists <- dist(t(assay(rld)))
- library("pheatmap")
- library("RColorBrewer")
- sampleDistMatrix <- as.matrix( sampleDists )
- colors <- colorRampPalette( rev(brewer.pal(9, "Blues")) )(255)
- pheatmap(sampleDistMatrix,
- clustering_distance_rows = sampleDists,
- clustering_distance_cols = sampleDists,
- col = colors)
同样的可以用Poisson Distance (Witten 2011)计算距离,计算方式如下:
- library("PoiClaClu")
- poisd <- PoissonDistance(t(counts(dds)))
PCA分析
还有一种可视化样本-样本距离的方法就是主成分分析。PCA分析我打算找点资料好好理解之后再写,这个说下有这个方法。
DESeq2提供了专门的方法用于作图,还很好看呢!
- plotPCA(rld,intgroup=c("condition"))
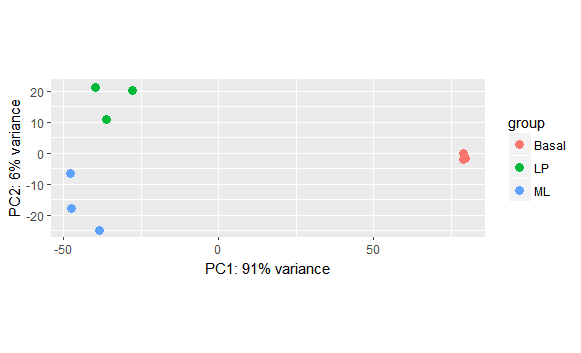
能够明显的发现不同处理的距离离得很远。
差异表达基因分析(DEA)
在我们定义好DESeqDataSe 就可以方便的调用DESeq分析.
- dds <- DESeq(dds)
在几行输出后信息后,分析就完成了,更多具体参数可以用?DESeq查看手册
构建结果表格
如果直接调用results,只会提取出design matrix最后两个变量(这里是ML vs Basal)的 log2 fold changes and p values等结果。
- > results(dds)
- log2 fold change (MAP): condition ML vs Basal
- Wald test p-value: condition ML vs Basal
- DataFrame with 27179 rows and 6 columns
- baseMean log2FoldChange lfcSE stat pvalue padj
- <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
- 497097 118.7522256 -7.0662875 0.5802473 -12.1780608 4.068401e-34 8.556128e-33
- 100503874 1.3308906 -2.6371037 1.3370584 -1.9723174 4.857338e-02 8.946685e-02
- 100038431 0.0843771 -0.1541107 1.2443948 -0.1238439 9.014388e-01 NA
- 19888 1.4833878 2.9503142 1.3351543 2.2097179 2.712475e-02 5.385925e-02
- 20671 26.4317752 -1.5256766 0.7754853 -1.9673829 4.913908e-02 9.034136e-02
- ... ... ... ... ... ... ...
- 100861837 368.84237 0.33334299 0.3226665 1.0330883 0.3015626 0.4139846
- 100861924 22.79272 -1.25169702 0.8307249 -1.5067528 0.1318740 0.2107990
- 170942 852.61538 -0.07612745 0.2399318 -0.3172879 0.7510252 0.8235139
- 100861691 0.00000 NA NA NA NA NA
- 100504472 0.00000 NA NA NA NA NA
实际上results有如下众多参数
- results(object, contrast, name, lfcThreshold = 0,
- altHypothesis = c("greaterAbs", "lessAbs", "greater", "less"),
- listValues = c(1, -1), cooksCutoff, independentFiltering = TRUE,
- alpha = 0.1, filter, theta, pAdjustMethod = "BH", filterFun,
- format = c("DataFrame", "GRanges", "GRangesList"), test, addMLE = FALSE,
- tidy = FALSE, parallel = FALSE, BPPARAM = bpparam(), ...)
比如可以指定比较对象Basal和PL,可以用mcols查看结果存储的元数据,了解列名的含义。
- res <- results(dds,contrast = c("condition","Basal","LP"))
- mcols(res, use.names = TRUE)
- DataFrame with 6 rows and 2 columns
- type description
- <character> <character>
- baseMean intermediate mean of normalized counts for all samples
- log2FoldChange results log2 fold change (MAP): condition Basal vs LP
- lfcSE results standard error: condition Basal vs LP
- stat results Wald statistic: condition Basal vs LP
- pvalue results Wald test p-value: condition Basal vs LP
- padj results BH adjusted p-valuess
其中lfcSE是Log2 fold change的标准误。其他项都比较好理解。
最后还可以看一些总结性的内容
- summary(res)
- out of 22026 with nonzero total read count
- adjusted p-value < 0.1
- LFC > 0 (up) : 5528, 25%
- LFC < 0 (down) : 5274, 24%
- outliers [1] : 43, 0.2%
- low counts [2] : 2953, 13%
- (mean count < 1)
- [1] see 'cooksCutoff' argument of ?results
- [2] see 'independentFiltering' argument of ?results
在limma分析结果分别是4127,4298,稍微多了那么2000个基因。其他结果也基本上都了2000个。看起来对于22000多个基因而言,差异不算太大。
当然我们还可以通过
- 降低假阳性率(FDR, false discovery rate)阈值
- 提高log2 fold change的阈值
- > res0.5 <- results(dds, contrast = c("condition","Basal","LP"),alpha=0.05)
- > table(res0.5$padj < 0.05)
- FALSE TRUE
- 8860 9750
- > resLFC1 <- results(dds, lcontrast = c("condition","Basal","LP"),fcThreshold=1)
- > table(resLFC1$padj < 0.1)
- FALSE TRUE
- 8916 10958
于是乎结果就和limma差不多了。
多重试验
在高通量数据分析中,我们通常不是用p值来拒绝原假设,更多是用来进行多重试验矫正。
为什么要做多重试验矫正?
如果对一个基因,我有99%的把握认为是差异表达的,也就是说1%的可能是错的。那么假设有10000个基因,按照数学期望,有100个是假的。因此为了保证多重试验结果的可靠性要对结果的p-value做矫正。
DESeq2用的Benjamini-Hochberg (BH) adjustment (Benjamini and Hochberg 1995),计算矫正后的P-value,用于回答如下问题:
如果我们选择了所有小于或等于矫正p-value阈值的所有显著性基因,假阳性比例( false discovery rate, FDR)是多少?
FDR在高通量试验中是比较有用的统计值,由于我们通常关注一类自己感兴趣的基因,所以我们需要设置一个假阳性上限。
我们从结果中以1%作为显著性,分别找出显著性上调和下调的基因。
- regSig <- subset(res, padj < 0.01)
- # Up
- head(resSig[ order(resSig$log2FoldChange), ])
- # Down
- head(resSig[ order(resSig$log2FoldChange, decreasing = TRUE), ])
可视化
人类对图像比较敏感,对文字比较迟钝。所以我们需要一些比较好看的图来放到文章中解释说明。
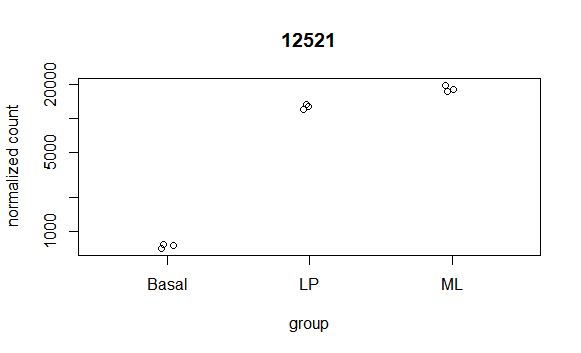
最简单的就是Counts plot,看看特定基因的count数量。
- topGene <- rownames(res)[which.min(res$padj)]
- plotCounts(dds, gene = topGene, intgroup=c("condition"))
可以用MA-plot(也叫mean-difference plot,Bland-Altman plot)了解模型(如所有基因在不同处理比较的结果)的估计系数的分布
- res <- lfcShrink(dds, contrast=c("contion","ML","PL"), res=res)
- plotMA(res, ylim = c(-5, 5))
更加喜闻乐见的是基因聚类所提供的热图展示。我们可以找前20个样本件差异比较大,然后看他们在不同样本间的表达情况。
- library("genefilter")
- topVarGenes <- head(order(rowVars(assay(rld)), decreasing = TRUE), 20)
- mat <- assay(rld)[ topVarGenes, ]
- mat <- mat - rowMeans(mat)
- anno <- as.data.frame(colData(rld)[, c("condition","lane")])
- pheatmap(mat,annotation_col = anno)
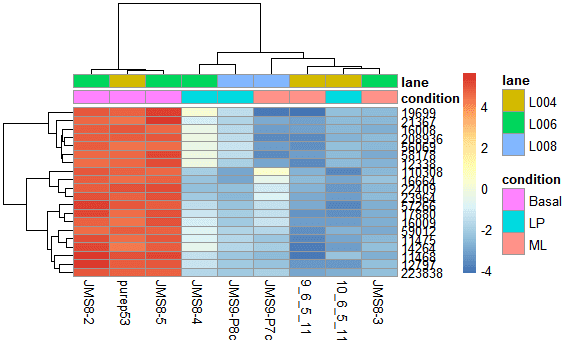
大致可以发现同一组的基因颜色是相同的,也就是说表达量相近。
结语
找到差异表达的基因只是第一步,后续还需要对这些基因进行进一步的分析,如
- 基因富集分析
- 主成分分析
- 聚类分析