

今天给大家推荐一个高通量测序数据质控神器——Trimmomatic。这个于 2014 年发表在 Bioinformatics 上的软件,至今为止在 Web of Science 上可以检索到 2,098 次引用,而在谷歌学术上更是达到了惊人的 3,391 次:

这个软件为什么深受大家的喜爱呢?今天小编就给大家分析一下它在质控方面的强大之处。
一 无脑安装、使用"简单"、运行速度可观
软件的安装方法请见:没有root管理员权限安装常用群体遗传学分析软件,使用方法可参考:Illumina测序数据的质量控制,本文在最后也会给出详细的说明。这个软件是用 Java 写的,运行效率比较高,本次测试用的机器 CPU 信息为:
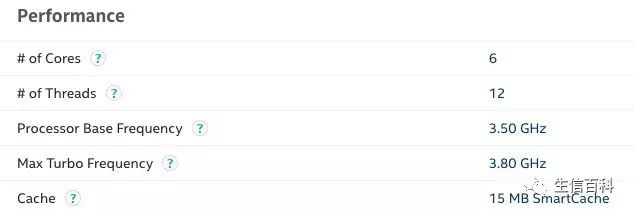
使用 4 个线程进行计算,处理一个 lane 的 raw data (Hiseq X ten,144.79 G 数据量) 需要 5 个小时20分钟,增加使用的线程数可以明显地加快运行速度。
二 强大的去接头能力
一般的质控软件在处理含有接头序列的 reads 时,通常采用 "在允许错配的情况下,如果分析的 read 匹配一定数量的接头序列即去除这条 read 或从匹配开始的位置截断 read,仅保留匹配位置之前的部分序列" 的方式。
- 如果采取 "去除含有接头序列的 reads" 的方式,会造成测序数据的浪费 (如果片段选择没有控制好,整个 lane 会有很大一部分数据含有接头序列,怎么办?);
- 如果采取 "从匹配开始的位置截断 read,仅保留匹配位置之前的部分序列" 的方式,对于只含有少数几个碱基的 reads,普通的质控软件是处理不了的(又该怎么办?)。
But,Trimmomatic 有两种模式:Single End Mode 和 Paired End Mode,对于单端测序数据,它和其它软件相比没有明显的优势;但如果是双端测序的数据,Trimmomatic 采用两种去接头方式,更强大,更彻底!
- 普通模式:匹配一定数量的接头序列即截断序列,保留匹配起始位置之前的序列,如下图中A、B 所示:
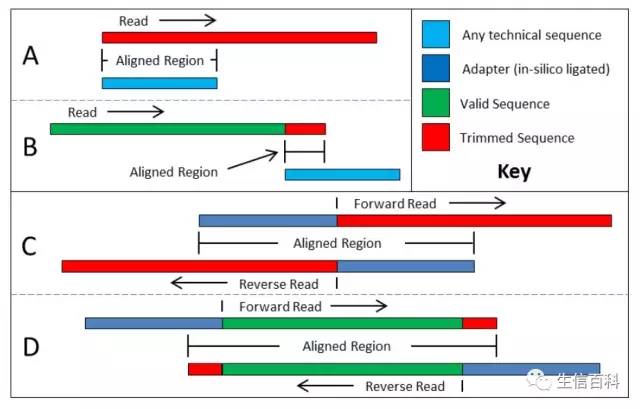
A、如果从 reads 的开始就匹配到接头序列的话,整条 reads 会被去除;
B、如果是从 reads 的其它部分匹配到接头序列,则从匹配的位置截断序列,保留包含接头的部分。
- 超级强大的回文模式,如上图 C和 D 所示:想要了解回文模式去接头的原理,我们需要先熟悉一下:测序结果中的接头序列来自哪里? 由于只有当插入片段的长度小于测序的读长时才会在测序结果中出现接头序列。那么对于含有接头的片段,正反向的 reads 在除接头之外的部分应该是反向互补的。因此,对于双端测序数据的处理上,Trimmomatic 在考虑接头匹配情况的同时也检查正反向 reads 的序列,从而更加有效的去掉接头序列。理论上,即使 read 仅含有 1 个碱基的接头序列,这 1 个碱基也能被切除!
需要了解更详细的信息,请查阅软件的说明书:http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/TrimmomaticManual_V0.32.pdf
三 多种低质量数据处理方式
- 可以使用 LEADING 和 TRAILING 参数,从序列的开头或者末尾截掉碱基质量值小于特定值的碱基;
- 可以使用 CROP 和 HEADCROP 参数,从序列的开头或者末尾截截取一定数目的碱基;
- 可以使用 SLIDINGWINDOW 方式,从 5' 端开始以设定的窗口大小计算碱基平均质量,如果此平均值低于设定的阈值,则从这个位置截断 read,这个模式对于整体测序质量很好但 reads 内含有连续 N 的情况很适用;
- 设置 AVGQUAL 参数,如果整条 read 的碱基平均质量值低于设定的阈值则去掉整条 read;
- 设置 MINLEN 参数,如果 read 长度低于设定的阈值则去掉整条 read;
- MAXINFO 同时考虑 reads 长度和错误率来进行质控。
四 更人性化
有些质控软件在处理双端测序数据时,得到的 clean data 不再是一一对应的 pair-end 形式,需要自己写代码或者找其它软件处理后,才能用于下一步的 mapping。 Trimmomatic 很好的解决来这种情况,如下图所示:
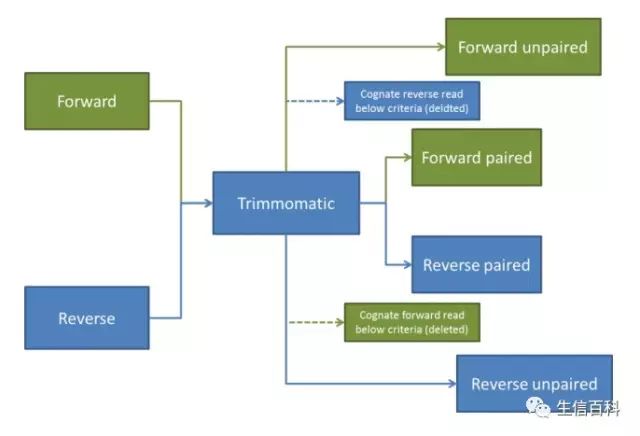
它含有 2 个输入文件 (配对的 pair-end 数据);4个输出文件,成对的 clean data, 未成对的正向序列以及未成对的反向序列,可以很方便的用于接下来的 mapping。
分析代码及解释
- Trimmomatic=/your/path/to/Trimmomatic-0.36/trimmomatic-0.36.jar
- adapter=/your/path/to/adapters
- java -jar "$Trimmomatic" PE -phred33 ${name}_1.fq.gz ${name}_2.trim.fq.gz ${name}_R1.clean.fq.gz ${name}_R1.unpaired.fq.gz ${name}_R2.clean.fq.gz ${name}_R2.unpaired.fq.gz ILLUMINACLIP:"$adapter"/Exome.fa:2:30:10:1:TRUE LEADING:20 TRAILING:20 SLIDINGWINDOW:4:15 -threads 2 MINLEN:36
其中,${name}_1.fq.gz ${name}_2.trim.fq.gz 为输入文件;
${name}_R1.clean.fq.gz ${name}_R1.unpaired.fq.gz ${name}_R2.clean.fq.gz ${name}_R2.unpaired.fq.gz 为对应的四个输出文件;
ILLUMINACLIP:"$adapter"/Exome.fa:2:30:9:1:TRUE,这部分指定 2 种去接头模式的参数:"$adapter"/Exome.fa 指明需要匹配的接头文件,2 代表 16 个碱基长度的种子序列中可以有 2 个错配,30 代表采用回文模式时匹配得分至少为30 (约50个碱基),10 代表采用简单模式时匹配得分至少为10 (约17 个碱基);
LEADING:20,从序列的开头开始去掉质量值小于 20 的碱基;
TRAILING:20,从序列的末尾开始去掉质量值小于 20 的碱基;
SLIDINGWINDOW:4:15,从 5' 端开始以 4 bp 的窗口计算碱基平均质量,如果此平均值低于 15,则从这个位置截断 read;
MINLEN:36, 如果 reads 长度小于 36 bp 则扔掉整条 read。
1F
学习了
2F
高通量测序数据质控神器—Trimmomatic