

我们先来看一下不同平台数据格式:
1. 产出数据格式
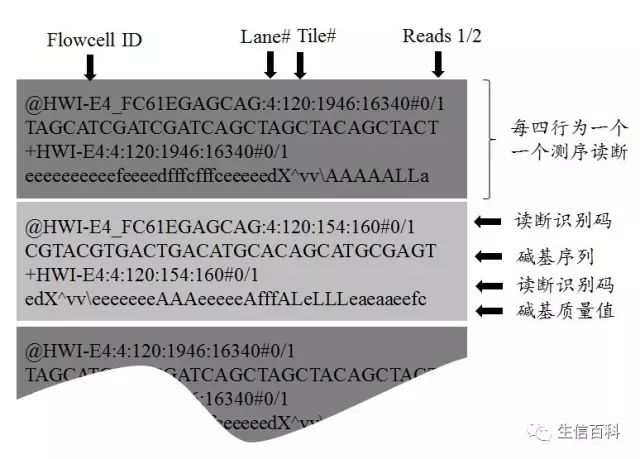
Solexa—fastq
2. 产出数据格式
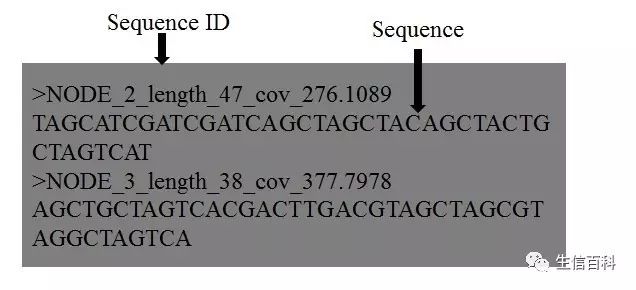
454—fasta
测序读长即每条测序读断的长度,所有平时大家叫的reads就是测序仪产生的读断。当然目前基因组拼接的主流平台仍然是Illumina,那么什么叫做测序数据质量值呢?质量值即每个碱基测序的错误率,如果p表示错误的可能性,那么质量值与这种可能性如何对应呢:
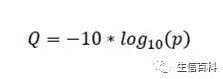
下图为错误率分别是0.1、0.01、0.001、0.0001时的碱基质量值的大小。
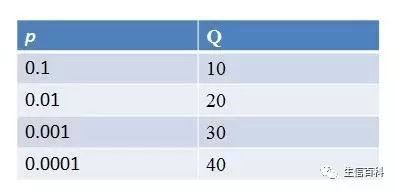
当然,测序碱基的错误率越小,质量值越高。所以如果你看到你的测序结果Q30的 reads数占到总Reads数的98%以上,那么恭喜你,这次测序非常好。
小伙伴表示:我明明看到reads质量值一行都是一些乱码,哪来的质量值?是这样的,测序仪产出的质量值都是一些ASCII码,也就是键盘上的十进制数,需要将其转换为16进制并减去Qphred的结果即为该碱基的测序质量值,按照测序时的设定值进行选择33或者64。
我们来看一下reads碱基测序错误率分布:
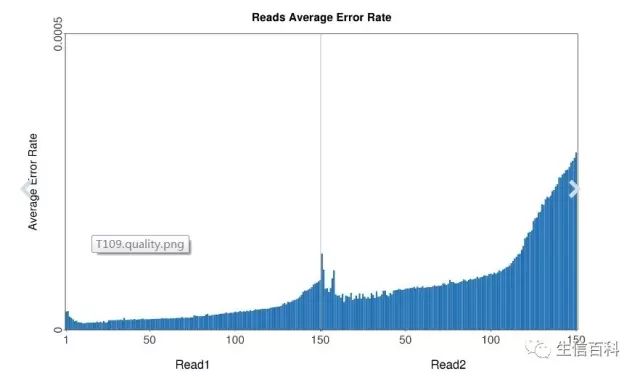
也就是说随着测序的进行,测序的错误率是随之升高的,这可能是由于聚合酶的保真度降低,二代测序固有的特点导致(荧光标记基团)。
以下为碱基分布类型的分布图:
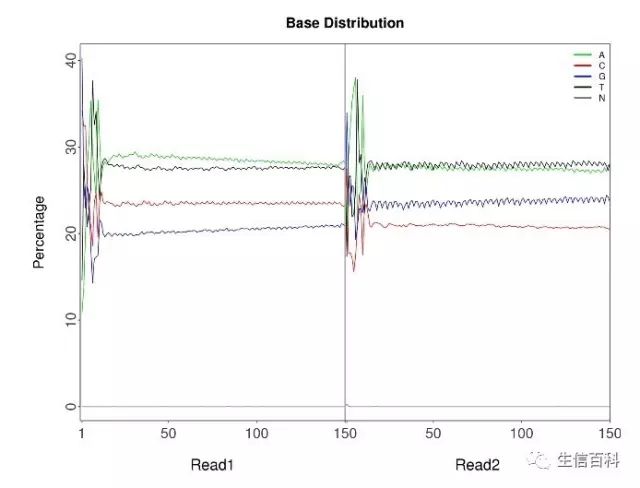
碱基类型分布检查用于检测有无AT、GC分离现象,由于所测的序列为随机打断的DNA片段,因随机性打断及碱基互补配对原则,理论上,G和C、A和T的含量在每个测序循环中应分别相等,长链非编码测序多采用链特异建库,GC、AT会存在一定的波动。由于Reads 5’端的前几个碱基为随机引物序列,存在一定的偏好性,因此会在碱基分布图中出现前端波动较大的现象。
那么,我们怎样对我们的数据进行质量评估与过滤呢。
大部分公司给的都是Clean data,也就是高质量的reads,如果不幸你拿到的是rawdata,或者自己测序的数据怎么办?
Trimmomatic是一个针对 Illumina数据处理工具。可针对paired-end 也能处理single ended。它能够利用fastq文件(phred + 33或者phred + 64碱基质量格式,取决于Illumina测序的机器)。
Trimmomatic用两种策略来去除adapter: Palindrome and Simple
软件下载地址:
http://www.usadellab.org/cms/?page=trimmomatic
PairedEnd Mode:
- java -jar <path totrimmomatic.jar> PE [-threads <threads] [-phred33 | -phred64] [-trimlog<logFile>] <input 1> <input 2> <paired output 1><unpaired output 1> <paired output 2> <unpaired output 2><step 1> ...
- # or
- java -classpath <path to trimmomaticjar> org.usadellab.trimmomatic.TrimmomaticPE [-threads <threads>][-phred33 | -phred64] [-trimlog <logFile>] <input 1> <input2> <paired output 1> <unpaired output 1> <paired output 2><unpaired output 2> <step 1> ...
SingleEnd Mode:
- java -jar <path to trimmomatic jar> SE[-threads <threads>] [-phred33 | -phred64] [-trimlog <logFile>]<input> <output> <step 1> ...
- #or
- java -classpath <path to trimmomatic jar>org.usadellab.trimmomatic.TrimmomaticSE [-threads <threads>] [-phred33 |-phred64] [-trimlog <logFile>] <input> <output> <step1> ...
举例:

那么什么样的结果才是合格的测序结果呢?
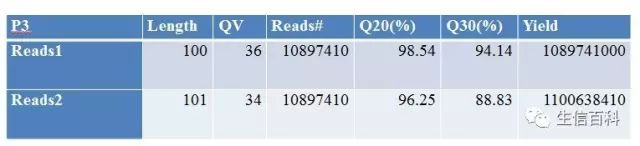
如果说Q20的reads可以达到95%以上,Q30的reads达到85%以上,我们认为本次测序还可以。当然我们可以通过过滤提高高质量reads的比率,当然测序量需达到要求。