

声明:本文为QIIME2官方帮助文档的中文版,由中科院遗传发育所刘永鑫博士翻译并亲测有效,文档翻译己获QIIME2团队官方授权。由于QIIME2更新频繁,如使用中遇到问题请访问QIIME2官方论坛阅读最新版中文帮助。
如中文翻译没有及时更新,新阅读英文原版 https://docs.qiime2.org68
本人只习惯使用命令行模式分析数据,图形界面和Ipython模式下使用暂不介绍。本系列的教程主要以命令行方式为大家演示。在其它方面有使用的经验的朋友,欢迎共享您的使用笔记发布于“宏基因组”公众号,方便大家学习和使用。
简介
QIIME2是微生物组分析流程QIIME(截止17.7.13被引7771次)的全新版(不是升级版),采用python3全新编写,并于2018年1月全面接档QIIME,是代表末来的分析方法标准(大牛们制定方法标准,我们跟着用就好了)。
优点
- 更易于安装:曾经QIIME的安装让无数生信人竞折腰,QIIME2引入了Miniconda软件包管理器,没有管理员权限也可以轻松安装;同时发布了docker镜像,下载即可运行;
- 使用方法多样:支持命令行模式(q2cli),也支持图型用户界面q2studio;还有Python用户喜欢的Artifact API(类似IPython notebook);
- 分析流程化:分析流程更加标准化,不让用户盲然下面该做什么;
- 可视化增强:QIIME后发制人,超越引用6964次的mothur流程,就是其可视化方面的优势,现可视化结果更加漂亮,且全新采用交互式图形结果,点选可查看细节,更易于分析;
- 方便合作:项目很少一个组可完成,多人多地结果图表方便共享,适合当下科研合作的需求;
- 可扩展:支持自定义功能并加入分析流程;高手可以自己写包,加入QIIME2的流程中了;
- 分析可重复:全新定义了文件系统,即包括分析数据、也包括分析过程和结果,每一步的结果,均可追溯全部分析过程,方便检查和重复。
安装
有多种安装方法,根据环境任选其一即可,先后序列即推荐序列。
1. Miniconda软件包管理器安装(需要有Linux服务器,但无需管理员权限)
本人测试采用Miniconda安装QIIME2于Ubuntu 16.04,这也是官方推荐的方法,确实非常简单。
# 安装miniconda软件管理器:用于安装QIIME2及依赖关系 https://conda.io/miniconda.html
wget https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh
chmod +x Miniconda3-latest-Linux-x86_64.sh
./Miniconda3-latest-Linux-x86_64.sh
# 按提示操作,如回车确定,输入yes同意许可,输入安装目录或回车按默认安装;
# 完成后关闭当前终端,新打开一个终端继续操作才能生效
# 升级conda为最新版:新版的bug最少,碰到问题的机会也小
conda update conda
# 安装qiime2-2017.7,下载安装所有依赖关系,网速不没,时间差别很大,我安装大约半小时
conda create -n qiime2-2017.7 --file https://data.qiime2.org/distro/core/qiime2-2017.7-conda-linux-64.txt
# 激活工作环境,需要几十秒
source activate qiime2-2017.7
# 检查是否安装成功,弹出程序帮助即成功
qiime --help
# 关闭工作环境:不用时关闭,不然你其它程序可能会出错
source deactivate
2. Docker方式安装,Linux需要管理员权限
我比较喜欢使用docker,直接下载预配置好的系统使用,对本地系统无影响
Dokcer的基本操作请查看本公众号之前的教程扩增子分析流程2.使用Docker运行QIIME34,或后台回复docker;
# 下载2017年6月最新版QIIME2
docker pull qiime2/core:2017.7
# 运行QIIME2测试:显示成功
docker run -t -i -v $(pwd):/data qiime2/core:2017.7 qiime
# 启动docker命令行,挂载目录至data目录
docker run --rm -v $(pwd):/data --name=qiime -it qiime2/core:2017.7
这就相当于打开了一个软件工作环境,目录/data为当前工作目录,可方便分析数据
3. Windows 10 64x Pro + Docker
Docker已经出Windows版了,但是需要Windows10 64位专业版 + HyperV,或者Win7 64位以上+Docker toolsbox,我尝试了一下没成功,可能是我相关知识不足或环境不满足,想用Windows体验分析的可以尝试一下。
QIIME2版本升级
本文案以2016.6升级至2016.7为例
QIIME2目前只处于Alpha版,每月均有升级。如使用中存在问题,建议立马升级至最新版。本系统至2018年1月会推出Beat稳定版。
Linux conda安装版的升级
# 升级conda至最新版
conda update conda
# 直接安装现在的2017.7
conda install --file https://data.qiime2.org/distro/core/qiime2-2017.7-conda-linux-64.txt
# 加载2017.7的环境境
source activate qiime2-2017.7
# CondaEnvironmentNotFoundError: Could not find environment: qiime2-2017.7 .
Docker安装方式的升级
与其説是升级,其实是下载了一个全新的docker,但之前共用的文件还可以用。下载失败,也説找不到文件
docker pull qiime2/core:2017.7
# Tag 2017.7 not found in repository docker.io/qiime2/core
扩增子分析QIIME2. 2分析实战:人体不同部分微生物组Moving Pictures
本文的操作的前提是完成QIIME2的安装,想安装QIIME2请阅读《扩增子分析QIIME2. 1简介和安装》。
本示例的的数据来自文章《Moving pictures of the human microbiome》,Genome Biology 2011,取样来自两个人身体四个部位五个时间点,
启动QIIME2运行环境
对于上文提到了两种常用安装方法,我们每次在分析数据前,需要打开工作环境,根据情况选择对应的打开方式。
# 创建工作目录并进入
mkdir -p qiime2
cd qiime2
# Miniconda安装的请运行如下命令加载工作环境
source activate qiime2-2017.7
# 如果是docker安装的请运行如下命令,否则跳过此行
docker run --rm -v $(pwd):/data --name=qiime -it qiime2/core:2017.7
准备数据
# 创建并进入工作目录
mkdir -p qiime2-moving-pictures-tutorial
cd qiime2-moving-pictures-tutorial
# 下载实验设计,此文件位于google服务器,服务器无法翻墙的可以用windows翻墙下载并上传到工作目录;或尝试下载我建立的备份
wget -O "sample-metadata.tsv" "https://data.qiime2.org/2017.7/tutorials/moving-pictures/sample_metadata.tsv"
## 上面一步下载失败,可尝试删除空文件并使用我建立的备份链接下载;否则跳过下面两行命令
rm sample_metadata.tsv
wget http://bailab.genetics.ac.cn/markdown/sample-metadata.tsv
# 下载实验测序数据
mkdir -p emp-single-end-sequences
wget -O "emp-single-end-sequences/barcodes.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/moving-pictures/emp-single-end-sequences/barcodes.fastq.gz"
wget -O "emp-single-end-sequences/sequences.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/moving-pictures/emp-single-end-sequences/sequences.fastq.gz"
# 生成qiime需要的artifact文件(qiime文件格式,将原始数据格式标准化)
qiime tools import \
--type EMPSingleEndSequences \
--input-path emp-single-end-sequences \
--output-path emp-single-end-sequences.qza
拆分样品
# 按barcode拆分样品 Demultiplexing sequences
qiime demux emp-single \
--i-seqs emp-single-end-sequences.qza \
--m-barcodes-file sample-metadata.tsv \
--m-barcodes-category BarcodeSequence \
--o-per-sample-sequences demux.qza
# 结果统计
qiime demux summarize \
--i-data demux.qza \
--o-visualization demux.qzv
# 查看结果 (依赖XShell+XManager或其它ssh终端和图形界面软件)
qiime tools view demux.qzv
使用qiime tools view demux.qzv或访问 https://view.qiime2.org/?src=https%3A%2F%2Fdocs.qiime2.org%2F2017.7%2Fdata%2Ftutorials%2Fmoving-pictures%2Fdemux.qzv在线查看
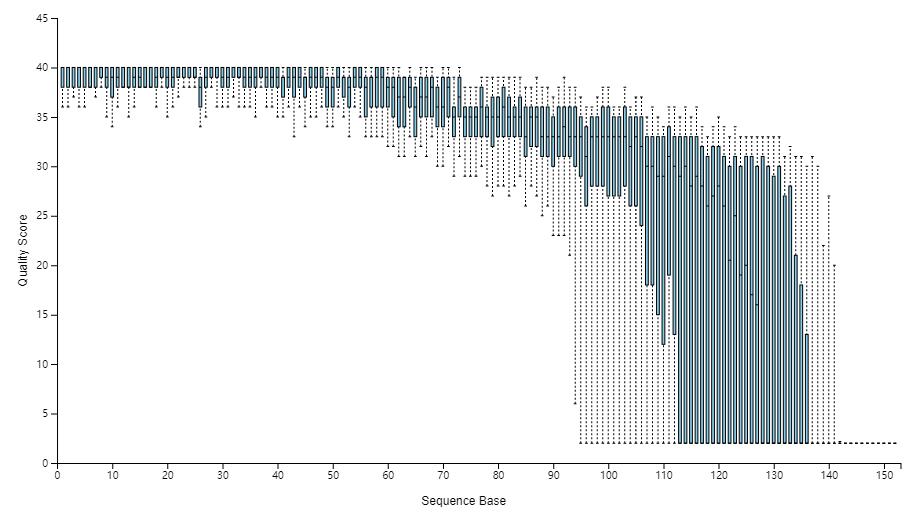
下图展示拆分样品的图表结果,其中的图表示各样品测序数据柱状分布图,展示不同测序深度下样品数量分布,可下载PDF文件;表可下载CSV格式文件,即每个样品的数据量表,可用Excel打开。

序列质控和生成OTU表
此步主要有DADA2和Deblur两种方法可选,推荐使用DADA2,去年发表在Nature Method上,比较同类方法优于其它OTU聚类结果;相较QIIME的UPARSE聚类方法,目前DADA2方法仅去噪去嵌合,不再按相似度聚类。比上一代分析结果更准确。
- DADA2
主要作用是去除低质量序列、嵌合体;再生成OTU表,现在叫Feature表,因为不再使用聚类方法,相当于QIIME时代100%相似度的OTU表。
读者思考时间:基于上图对拆分样品的统计结果,如何设置下面生成OTU表的参数。
- –p-trim-left 截取左端低质量序列,我们看上图中箱线图,左端质量都很高,无低质量区,设置为0;
- –p-trunc-len 序列截取长度,也是为了去除右端低质量序列,我们看到大于120以后,质量下降极大,甚至中位数都下降至20以下,需要全部去除。
# 单端序列去噪, 去除左端0bp(--p-trim-left用于切除边缘低质量区),序列切成120bp长;生成代表序列和OTU表;并重命名用于下游分析
qiime dada2 denoise-single \
--i-demultiplexed-seqs demux.qza \
--p-trim-left 0 \
--p-trunc-len 120 \
--o-representative-sequences rep-seqs-dada2.qza \
--o-table table-dada2.qza
mv rep-seqs-dada2.qza rep-seqs.qza
mv table-dada2.qza table.qza
- Deblur
与DADA2二选一,用户可自行比较结果的差异,根据喜好选择。
# 按测序质量过滤序列
qiime quality-filter q-score \
--i-demux demux.qza \
--o-filtered-sequences demux-filtered.qza \
--o-filter-stats demux-filter-stats.qza
# 去冗余生成OTU表和代表序列;结果文件名有deblur,没有用于下游分析,请读者想测试的自己尝试
qiime deblur denoise-16S \
--i-demultiplexed-seqs demux-filtered.qza \
--p-trim-length 120 \
--o-representative-sequences rep-seqs-deblur.qza \
--o-table table-deblur.qza \
--o-stats deblur-stats.qza
Feature表统计
qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv \
--m-sample-metadata-file sample-metadata.tsv
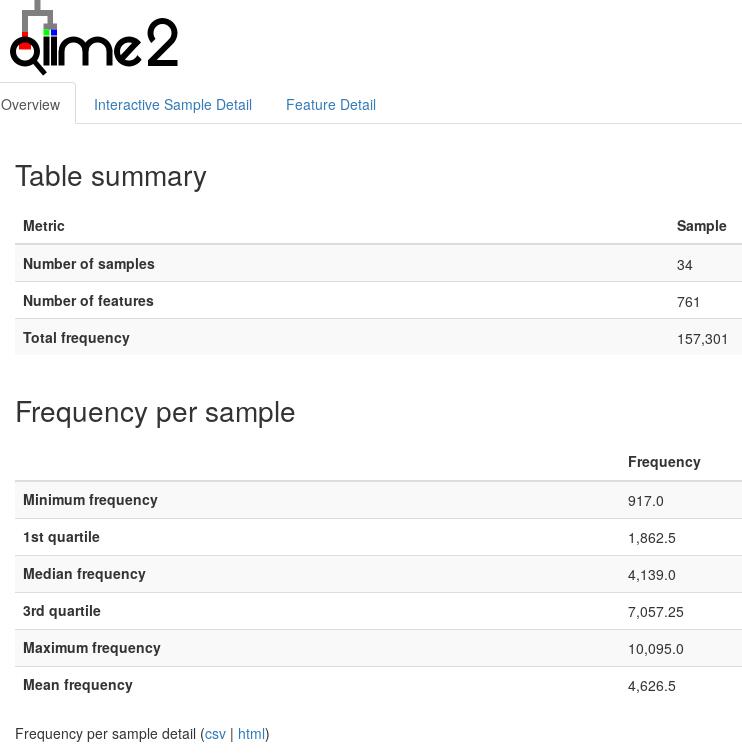
qiime tools view table.qzv # 展打开网页版统计结果,看下图
图中展示了Feature表的统计结果
代表序列统计
qiime feature-table tabulate-seqs \
--i-data rep-seqs.qza \
--o-visualization rep-seqs.qzv
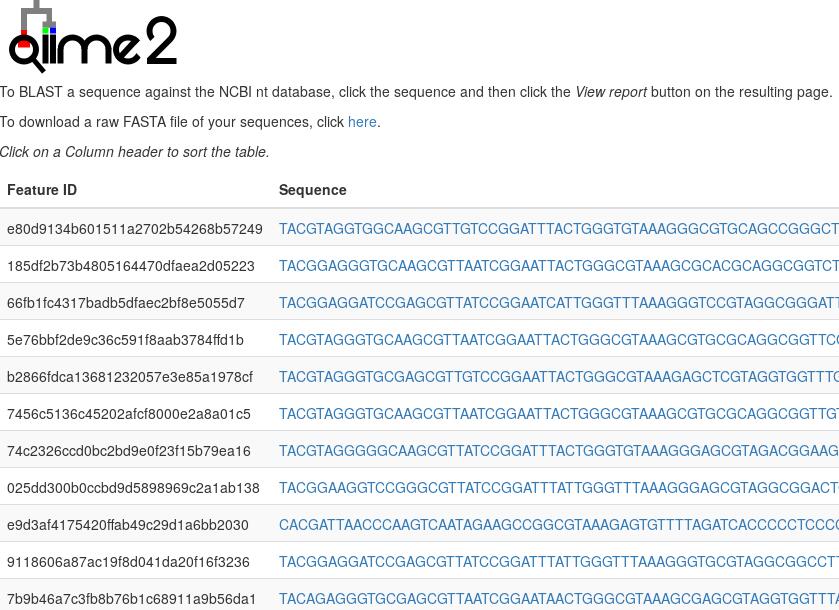
qiime tools view rep-seqs.qzv # 展打开网页版统计结果,看下图
下图展示代表序列网页版详细,点"Click here"可下载fasta文件
建树:用于多样性分析
# 多序列比对
qiime alignment mafft \
--i-sequences rep-seqs.qza \
--o-alignment aligned-rep-seqs.qza
# 移除高变区
qiime alignment mask \
--i-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza
# 建树
qiime phylogeny fasttree \
--i-alignment masked-aligned-rep-seqs.qza \
--o-tree unrooted-tree.qza
# 无根树转换为有根树
qiime phylogeny midpoint-root \
--i-tree unrooted-tree.qza \
--o-rooted-tree rooted-tree.qza
Alpha多样性
读者思考时间:下面多样性分析,需要基于标准化的OTU表,标准化采用重抽样至序列一致,如何设计样品重抽样深度参数。–p-sampling-depth
如是数据量都很大,选最小的即可。如果有个别数据量非常小,去除最小值再选最小值。比如此分析最小值为917,我们选择1080深度重抽样,即保留了大部分样品用于分析,又去除了数据量过低的异常值。
注:本示例为454时代的测序,数据量很小。现在一般采用HiSeq PE250测序,数据量都非常大,通常可以采用3万或5万的标准筛选,仍可保留90%以上样本。过低或过高一般结果也会异常,不建议放在一起分析。
# 计算多样性(包括所有常用的Alpha和Beta多样性方法),输入有根树、Feature表、样本重采样深度(一般为最小样本数据量,或覆盖绝大多数样品的数据量)
qiime diversity core-metrics \
--i-phylogeny rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth 1080 \
--output-dir core-metrics-results
# 输出结果包括多种多样性结果,文件列表和解释如下:
# beta多样性bray_curtis距离矩阵 bray_curtis_distance_matrix.qza
# alpha多样性evenness(均匀度,考虑物种和丰度)指数 evenness_vector.qza
# alpha多样性faith_pd(考虑物种间进化关系)指数 faith_pd_vector.qza
# beta多样性jaccard距离矩阵 jaccard_distance_matrix.qza
# alpha多样性observed_otus(OTU数量)指数 observed_otus_vector.qza
# alpha多样性香农熵(考虑物种和丰度)指数 shannon_vector.qza
# beta多样性unweighted_unifrac距离矩阵,不考虑丰度 unweighted_unifrac_distance_matrix.qza
# beta多样性unweighted_unifrac距离矩阵,考虑丰度 weighted_unifrac_distance_matrix.qza
# 统计faith_pd算法Alpha多样性组间差异是否显著,输入多样性值、实验设计,输出统计结果
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/faith_pd_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/faith-pd-group-significance.qzv
# 统计evenness组间差异是否显著
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/evenness_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/evenness-group-significance.qzv
# 网页展示结果,只要是qzv的文件,均可用qiime tools view查看或在线https://view.qiime2.org/查看,以后不再赘述
qiime tools view evenness-group-significance.qzv
以Evenness为例,下图展示多样性的箱线图,可以下载svg矢量图
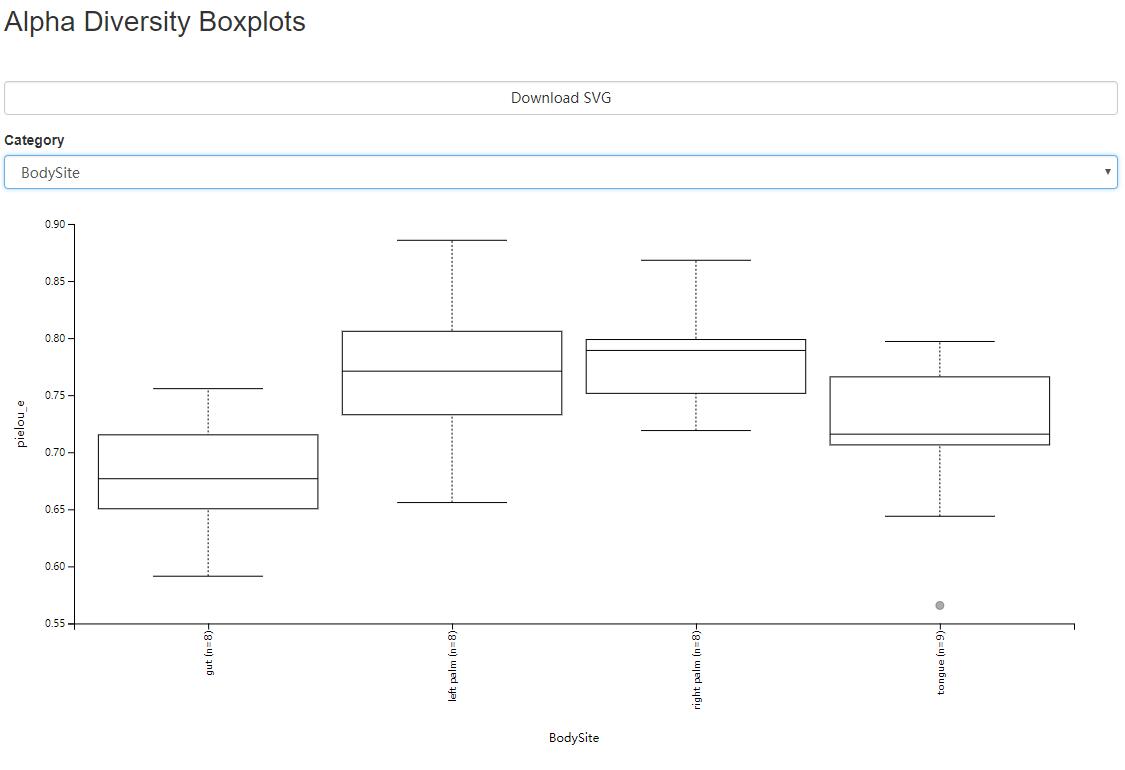
读者思考时间:实验设计中的那一种分组方法,与微生物群体的丰富度差异相关,这些差异显著吗?
解答:图中可按Catalogy选择分类方法,查看不同分组下箱线图间的分布与差别。图形下面的表格,详细详述组间比较的显著性和假阳性率统计。
结果我们会看到本实验设计的分组方式有Bodysite, Subject, ReportAntibioticUse,只有身体位置各组间差异明显,且下面统计结果也存在很多组间的显著性差异。
Beta多样性
# 按BodySite分组,统计unweighted_unifrace距离的组间是否有显著差异
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-category BodySite \
--o-visualization core-metrics-results/unweighted-unifrac-body-site-significance.qzv \
--p-pairwise
# 按Subject分组,统计unweighted_unifrace距离的组间是否有显著差异
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-category Subject \
--o-visualization core-metrics-results/unweighted-unifrac-subject-group-significance.qzv \
--p-pairwise
# 可视化三维展示unweighted-unifrac的主坐标轴分析
qiime emperor plot \
--i-pcoa core-metrics-results/unweighted_unifrac_pcoa_results.qza \
--m-metadata-file sample-metadata.tsv \
--p-custom-axis DaysSinceExperimentStart \
--o-visualization core-metrics-results/unweighted-unifrac-emperor.qzv
# 可视化三维展示bray-curtis的主坐标轴分析
qiime emperor plot \
--i-pcoa core-metrics-results/bray_curtis_pcoa_results.qza \
--m-metadata-file sample-metadata.tsv \
--p-custom-axis DaysSinceExperimentStart \
--o-visualization core-metrics-results/bray-curtis-emperor.qzv
# 网页展示结果,或下载在线查看
qiime tools view core-metrics-results/bray-curtis-emperor.qzv
以Bray_curtis可视化结果为例,下图展示多样性的三维散点图,可以下载svg矢量图

读者思考时间:按subject分组有显著区别吗?按body-site分组有显著区别吗?那些body-site组间存在区别?
大家仔细查看按subject进行主坐标轴分析的结果,如下链接,我是没看到区别;
https://view.qiime2.org/?src=https%3A%2F%2Fdocs.qiime2.org%2F2017.7%2Fdata%2Ftutorials%2Fmoving-pictures%2Fcore-metrics-results%2Funweighted-unifrac-subject-group-significance.qzv
大家仔细查看按body-site进行主坐标轴分析的结果,如下链接,只有左右手间无明显差别,其它各组间均有显著差别。
https://view.qiime2.org/?src=https%3A%2F%2Fdocs.qiime2.org%2F2017.7%2Fdata%2Ftutorials%2Fmoving-pictures%2Fcore-metrics-results%2Funweighted-unifrac-body-site-significance.qzv
按其它距离计算的结果,读者可以仔细看看不同距离矩阵计算结果的区别。个人感觉,一般比较好解释科学问题的方法就是适合的方法。
物种分类
# 下载物种注释
wget -O "gg-13-8-99-515-806-nb-classifier.qza" "https://data.qiime2.org/2017.7/common/gg-13-8-99-515-806-nb-classifier.qza"
# 物种分类
qiime feature-classifier classify-sklearn \
--i-classifier gg-13-8-99-515-806-nb-classifier.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
# 物种结果转换表格,可用于查看
qiime taxa tabulate \
--i-data taxonomy.qza \
--o-visualization taxonomy.qzv
# 展示taxonomy.qzv结果如下:
#Feature ID Taxonomy
#d12759fe8dda1d65fe9077cc1ca9cf28 k__Bacteria; p__Bacteroidetes; c__Flavobacteriia; o__Flavobacteriales; f__[Weeksellaceae]; g__Chryseobacterium; s__
#5ada68b9a081358e1a7d5f1d351e656a k__Bacteria; p__Fusobacteria; c__Fusobacteriia; o__Fusobacteriales; f__Leptotrichiaceae; g__Leptotrichia; s__
#d9095748835ade1b8914c5f57b6acbcf k__Bacteria; p__Proteobacteria; c__Gammaproteobacteria; o__Aeromonadales; f__Aeromonadaceae; g__Oceanisphaera; s__
# 物种分类柱状图
qiime taxa barplot \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization taxa-bar-plots.qzv
qiime tools view taxa-bar-plots.qzv
以堆叠柱状图,展示物种各组、各分类级别下的相对丰度,图形可交互查看每个柱的详细信息,也可以下载svg矢量图。
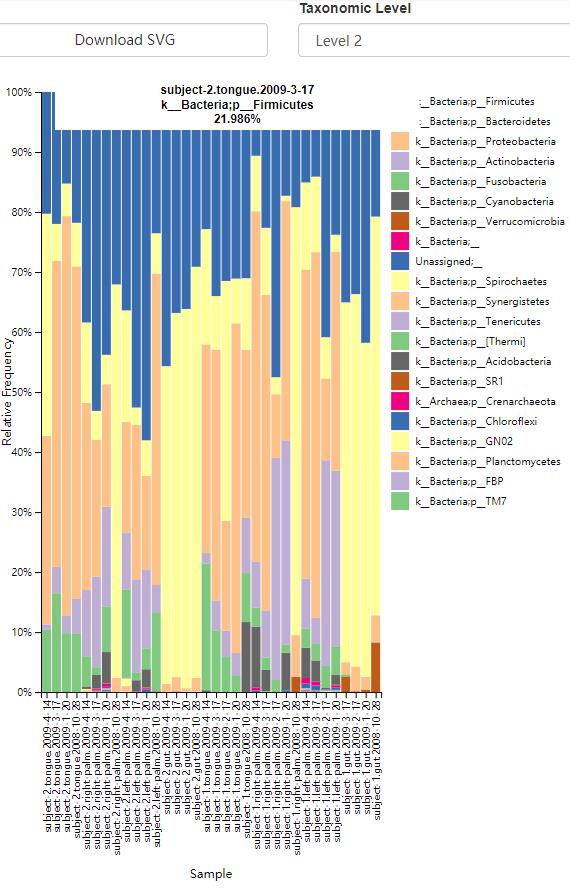
读者思考时间1:代表序列文件rep-seqs.qzv可视化结果中,可以下载fasta文件采用NCBI进行blast注释物种信息,与我们目前的结果比较,看看有什么不同,各分类级别的注释定义的相似程度是什么?
读者思考时间2:查看门水平(level2)分类结果柱状图,看每一类body-site中主要丰度的门类是什么?
差异丰度分析
差异丰度分析采用ANCOM (analysis of composition of microbiomes),是2015年发布在Microb Ecol Health Dis上的方法,文章称在微生物组方面更专业,但不接受零值(零在二代测序结果表中很常见)。我个人一直用edgeR,感觉靠谱,因为高通量测序本质上是相同的。
差异Features/OTUs分析
# OTU表添加假count,因为ANCOM不允许有零
qiime composition add-pseudocount \
--i-table table.qza \
--o-composition-table comp-table.qza
# 采用ancon,按BodySite分组进行差异统计
qiime composition ancom \
--i-table comp-table.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-category BodySite \
--o-visualization ancom-BodySite.qzv
# 查看结果
qiime tools view ancom-BodySite.qzv
读者思考时间:不同身体部分有那些Features存在丰度差异?那一组是最高或最低丰度?这此差异的Features属那些分类单元?
使用qiime tools view ancom-BodySite.qzv查看结果,或下载结果用qiime2网页在线打开查看,回答上面的问题。
差异分类学级别分析:以按门水平合并再统计差异
# 按门水平进行合并,统计各门的总reads
qiime taxa collapse \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--p-level 2 \
--o-collapsed-table table-l2.qza
# 同理去除零
qiime composition add-pseudocount \
--i-table table-l2.qza \
--o-composition-table comp-table-l2.qza
# 在门水平按取样部分分析
qiime composition ancom \
--i-table comp-table-l2.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-category BodySite \
--o-visualization l2-ancom-BodySite.qzv
可视化结果包括统计、各组丰度分位数和交互式火山图
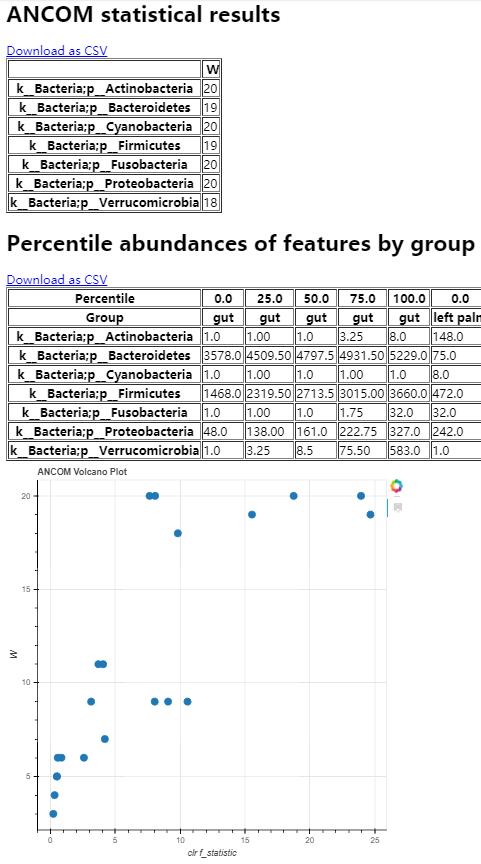
读者思考时间:不同身体部分有那些Features存在丰度差异?那一组是最高或最低丰度?这此差异的Features属那些分类单元?
使用qiime tools view ancom-BodySite.qzv查看结果,或点如下链接查看详细,回答上面的问题。
https://view.qiime2.org/?src=https%3A%2F%2Fdocs.qiime2.org%2F2017.7%2Fdata%2Ftutorials%2Fmoving-pictures%2Fl2-ancom-BodySite.qzv
结果描述:结果的可视化(Visual)页面,一共分为三部分。
- 第一个表为ANCOM statistical results,只列出组间存在显著差异的门,其统计值W的计算及解释尚不清楚,查原始文章也没有找到。有待更新版中解释。
- 第二个表为各组的丰度分位数,就是箱线图的原始数据,为什么作者没有直接出图,我将与作者沟通讨论;目前可以比较各组的分布,来具体看组间的差异,但不够直观;
- 统计各门类的火山图,坐标轴还没有详细解释,但其意思是越靠上越显著差异。此图采用Python的bokeh库生成的交互式图形,可以点击图中的点来查看具体的详细,如具体的分类学信息。相当于表1的可视化。
结果的网页还有其它页面,如peek页面可以查看此文件的基本信息,Provenance页面显示当前结果的生成过程图,点击过程中的点可以查看具体的程序和参数;链接按扭可以生成共享链接;下载按扭可以下载原始文件。

扩增子分析QIIME2. 3粪便菌群移植分析
原文地址: https://docs.qiime2.org/2017.7/tutorials/fmt/7
此实例需要一些基础知识,要求完成本系列文章前两篇内容:1简介和安装和2分析实战Moving Pictures。
本实验研究自闭症且胃肠道功能紊乱患者,采用粪便菌群移植方法,来降低患者的行为异常和肠道紊乱。监测移植后18个月范围内肠道菌群的变化,采用MiSeq PE300测序技术。
实验数据下载
# 激活工作环境
source activate qiime2-2017.7
# 创建工作目录并下载实验设计
mkdir qiime2-fmt-tutorial
cd qiime2-fmt-tutorial
wget -O "sample-metadata.tsv" "https://data.qiime2.org/2017.7/tutorials/fmt/sample_metadata.tsv"
# 下载成功跳过此步,下载失败处理方法:因为中国大陆用户没没有访问google的权限,请删除空文件,并下载备用链接
rm sample-metadata.tsv
wget http://bailab.genetics.ac.cn/markdown/QIIME2/sample-metadata.tsv
# 下载实验数据,为演示分析,只使用原始数据10%的抽样结果,如果下载失败请重试即可。
wget -O "fmt-tutorial-demux-1.qza" "https://data.qiime2.org/2017.7/tutorials/fmt/fmt-tutorial-demux-1-10p.qza"
wget -O "fmt-tutorial-demux-2.qza" "https://data.qiime2.org/2017.7/tutorials/fmt/fmt-tutorial-demux-2-10p.qza"
序列质控评估
# 序列质量评估
qiime demux summarize \
--i-data fmt-tutorial-demux-1.qza \
--o-visualization demux-summary-1.qzv
qiime demux summarize \
--i-data fmt-tutorial-demux-2.qza \
--o-visualization demux-summary-2.qzv
# 查看质量评估结果,用于确定下步质控的参数
qiime tools view demux-summary-1.qzv
qiime tools view demux-summary-2.qzv
查看可视化评估结果,也可下载qzv文件,使用 view.qiime2.org3 打开查看,或直接打开作者在线预计算好的结果。 https://view.qiime2.org/?src=https%3A%2F%2Fdocs.qiime2.org%2F2017.7%2Fdata%2Ftutorials%2Ffmt%2Fdemux-summary-1.qzv
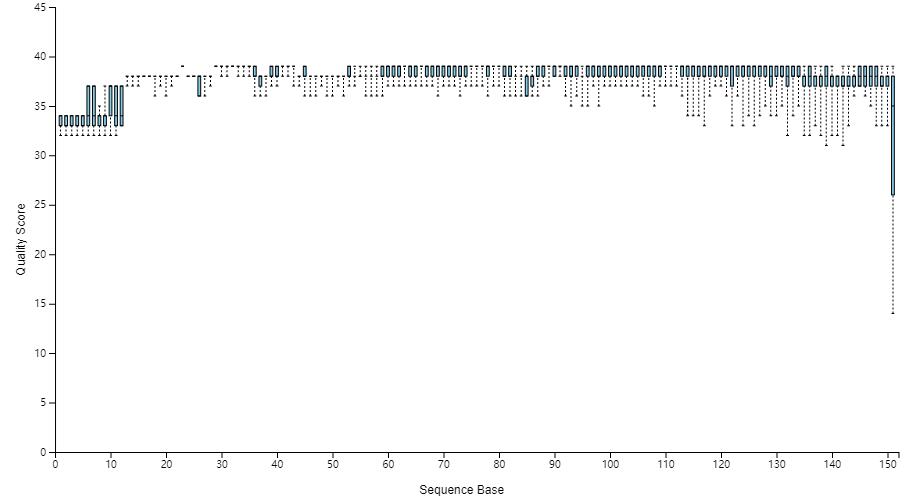
问题:从上图中我们判断选择质控的参数是什么?
序列上游13 bp的序列质量偏低,设置trim-left 13截掉前13bp序列;整体150bp的质量都不错,则保留150 bp的序列长度。
Feature表和代表性序列生成
# dada2去质控和去冗余,本实验有两批独立的数据,需要处理两次,生成代表序列和OTU表
qiime dada2 denoise-single \
--p-trim-left 13 \
--p-trunc-len 150 \
--i-demultiplexed-seqs fmt-tutorial-demux-1.qza \
--o-representative-sequences rep-seqs-1.qza \
--o-table table-1.qza
qiime dada2 denoise-single \
--p-trim-left 13 \
--p-trunc-len 150 \
--i-demultiplexed-seqs fmt-tutorial-demux-2.qza \
--o-representative-sequences rep-seqs-2.qza \
--o-table table-2.qza
合并不同组的序列和表
# 合并两组数据和OTU表
qiime feature-table merge \
--i-table1 table-1.qza \
--i-table2 table-2.qza \
--o-merged-table table.qza
# 合并两组数据的代表性序列
qiime feature-table merge-seq-data \
--i-data1 rep-seqs-1.qza \
--i-data2 rep-seqs-2.qza \
--o-merged-data rep-seqs.qza
# OTU表统计
qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv \
--m-sample-metadata-file sample-metadata.tsv
# 查看Feature/OTU表的统计结果
qiime tools view table.qzv
# 代表性序列统计
qiime feature-table tabulate-seqs \
--i-data rep-seqs.qza \
--o-visualization rep-seqs.qzv
# 查看代表性序列的统计结果
qiime tools view rep-seqs.qzv
table.qzv文件中Feature/OTU表统计的结果如下,或在线查看详细信息:https://view.qiime2.org/visualization/?type=html&src=https%3A%2F%2Fdocs.qiime2.org%2F2017.7%2Fdata%2Ftutorials%2Ffmt%2Ftable.qzv3
OTU表总结
| Metric | Sample |
|---|---|
| Number of samples | 121 |
| Number of features | 337 |
| Total frequency | 48,925 |
样品数据量分布
| Type | Frequency |
|---|---|
| Minimum frequency | 84.0 |
| 1st quartile | 276.0 |
| Median frequency | 380.0 |
| 3rd quartile | 492.0 |
| Maximum frequency | 860.0 |
| Mean frequency | 404.3388429752066 |
特征表频率统计
| Type | Frequency |
|---|---|
| Minimum frequency | 2.0 |
| 1st quartile | 9.0 |
| Median frequency | 24.0 |
| 3rd quartile | 85.0 |
| Maximum frequency | 10,832.0 |
| Mean frequency | 145.1780415430267 |
通过上表,我们可以确定Feature特征表标准化的重抽样数据,由于本测试,只用了文章原始数据的10%的数据,数据量很小,最小值为84,第一分位数为276,我们可选择276保留75%以上的样品。一般最小值最少1000,推荐5000以上。
多样性分析
现在我们已经获得了特征表(Feature/OTU),以及代表性序列(Feature Seq)
自己尝试用上篇文章的分析方法,回答下面这些问题。
前两节课的知识应该可以回答下面的问题,过两天我也会分析并做出答案。大家一定要实操才有真正分析的能力,想不出来就多操作和思路上一篇分析。
- 个人微生物组;
- 按subject-id分类存在组成差异?
- 按subject-id分类存在丰富度差异?
- 按subject-id分类存在均匀度差异?
- 菌群移植;
3. 移植几周后,患者的菌群在unweighted unifrac距离下最像供体;
4. 移植几周后,患者的菌群在bray-curtis距离下最像供体;
5. 比较两种距离结果那种解释更好; - 实验设计:比较粪便和试子样品采集方法
4. 比较不同取样方法结果中最大差别的类别?差异类别用blast,或classifier注释有什么不同?
5. 两类样品的unweighted unifrac和bray-curtis间有什么不同?
6. 供体粪便与那种取样的结果更像?
7. 两类取样方法的Alpha多样性存在差别吗? - 每个测序Run中有多少样品?在不同测序Run中是否存在系统性差异?
扩增子分析QIIME2. 4阿塔卡马沙漠微生物组分析
原文地址: https://docs.qiime2.org/2017.7/tutorials/atacama-soils/3
此实例需要一些基础知识,要求完成本系列文章前两篇内容:1简介和安装和2分析实战Moving Pictures。
本教程设计有两个目的:熟悉双端数据的处理;基于Moving Pictures后的主自分析练习以积累项目经验。
实验设计
本实验研究对像为智利北部的阿塔卡马沙漠。 此地为世界上最干旱的地区之一,其中有些地方十年降水量不足1毫米。尽管这里极端干旱,但仍有微生物生活在这里。我们的采样地点为东部的Baquedano和西部的Yungay,发现土壤温度与降水量正相关。在这两个地点,我们挖坑,并在不同深度取三组样品。
实验数据下载
# 激活工作环境
source activate qiime2-2017.7
# 建立工作目录
mkdir qiime2-atacama-tutorial
cd qiime2-atacama-tutorial
# 下载实验设计:Google文档在中国大陆无法下载,直接使用我建立的备用链接
# wget -O "sample-metadata.tsv" "https://data.qiime2.org/2017.7/tutorials/atacama-soils/sample_metadata.tsv"
wget http://bailab.genetics.ac.cn/markdown/QIIME2/atacama-soils/sample-metadata.tsv
# 下载双端实验数据(使用10%抽样数据方便下载和演示):分别为正向、反向和barcodes序列三个文件;文来自亚马逊云,有时无法下载或断开,只下载一个文件不同时间多试几次就成功了。
mkdir emp-paired-end-sequences
wget -O "emp-paired-end-sequences/forward.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/atacama-soils/10p/forward.fastq.gz"
wget -O "emp-paired-end-sequences/reverse.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/atacama-soils/10p/reverse.fastq.gz"
wget -O "emp-paired-end-sequences/barcodes.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/atacama-soils/10p/barcodes.fastq.gz"
双端数据分析方法
# 双端数据导入,数据建库类型为EMP双端序列(本示例来自EMP项目)
qiime tools import \
--type EMPPairedEndSequences \
--input-path emp-paired-end-sequences \
--output-path emp-paired-end-sequences.qza
# 按Barcode进行样品:--m-barcodes-file为含有样品与barcode信息对应的实验设计,--m-barcodes-category指定含有barcode信息的列名称,--i-seqs输入文件,--o-per-sample-sequences输出文件, --p-rev-comp-mapping-barcodes为barcode方向,是用实验设计的barcode与测序文件比对确定方向,此分析中为反向互补
qiime demux emp-paired \
--m-barcodes-file sample-metadata.tsv \
--m-barcodes-category BarcodeSequence \
--i-seqs emp-paired-end-sequences.qza \
--o-per-sample-sequences demux \
--p-rev-comp-mapping-barcodes
# 对拆分样品的结果和质量进行统计
qiime demux summarize \
--i-data demux.qza \
--o-visualization demux.qzv
# 展示统计分析结果
qiime tools view demux.qzv
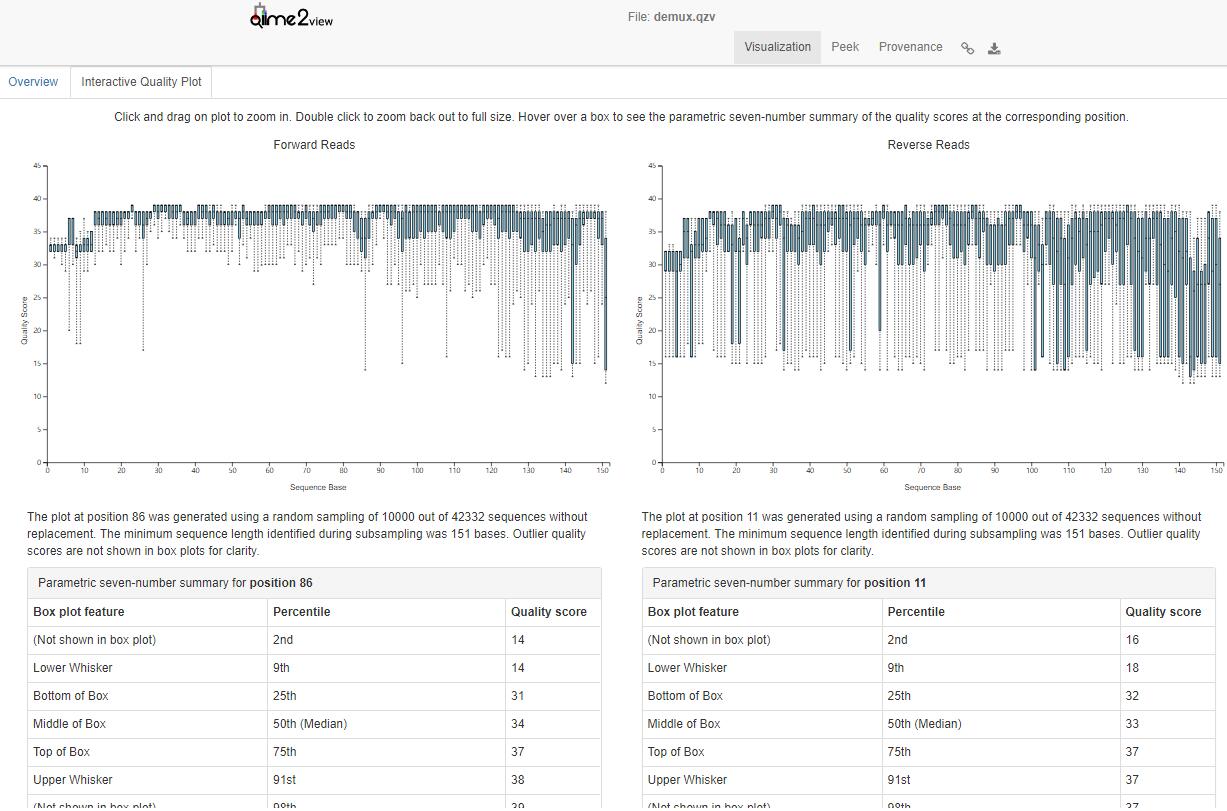
质量分析后,我们根据上图结果和相关表格来确定下步denoise分析参数。详细信息查看点下面链接
https://view.qiime2.org/visualization/?type=html&src=https%3A%2F%2Fdocs.qiime2.org%2F2017.7%2Fdata%2Ftutorials%2Fatacama-soils%2Fdemux.qzv6 。网页中交互式图形可以查看每个碱基位置的详细信息。
去噪并生成Feature表和代表性序列
设置左端截掉13bp,右端不截。(我通常处理是先合并双端,再质控,再去引物,虽然复杂更感觉更合理。但此方法可能更适合EMP项目,每个人的设计不同,参数不同)
qiime dada2 denoise-paired \
--i-demultiplexed-seqs demux.qza \
--o-table table \
--o-representative-sequences rep-seqs \
--p-trim-left-f 13 \
--p-trim-left-r 13 \
--p-trunc-len-f 150 \
--p-trunc-len-r 150
# 查看Feature/OTU表的统计结果
qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv \
--m-sample-metadata-file sample-metadata.tsv
# 代表性序列统计
qiime feature-table tabulate-seqs \
--i-data rep-seqs.qza \
--o-visualization rep-seqs.qzv
结果本地qiime tools view查看,本地没有配置可视化环境的,请下载文件在线查看 view.qiime2.org3
大家先本地或在线分析Feature表和代表性序列的统计结果,先熟悉数据。再按照2分析实战中方法,继续建树、多样性分析及比较,回答下面的问题?
接下来分析并回答的问题
- 接下来OTU表标准化参数–p-sampling-depth应该选多少?有多少样品应该从实验中剔除?过滤后核心矩阵中有多少数据量?
- 实验设计中的那种分级方式中微生物组成差异最大?采用那种距离计算方法分开更明显,是unweighted UniFrac还是Bray-Curtis?考虑尝试使用
qiime diversity beta-correlation and qiime diversity bioenv分析结果,可以使用--help查看详细帮助。 - 使用
qiime diversity alpha-correlation分析样品间的相关性,看看能得到什么结论? - 按组分析Alpha多样性,并比较是否有显著差异?
- 在门水平查看不同温度下微生物组成?看那些种类与湿度相关?
- 在有无植被的取样地点,什么菌门差异明显?
Acknowledgements
本文的数据来自 干旱土壤微生物组:显著增加与温度,在mSystems正在审稿
扩增子分析QIIME2. 5数据导入Importing data
为什么要导入数据?
QIIME2使用了标准文件格式qza和qzv,分别是数据文件和统计图表文件;目的是统一文件格式,方便追溯分析过程。
本人将带大家熟悉QIIME2分析流程的不同阶段,导入数据。
最典型的导入数据,是原始测序数据的导入。实际上,我们可以从分析的任何一步导入数据,继续分析。比如合作者提供了biom格式的OTU表,我们可以导入,并进行下游的统计分析。
导入数据可以采用多种方式,包括命令行或图形界面,我们这里主要介绍命令行的方式。
# 安装QIIME2 2017.7,如己安装请跳过
conda update conda
conda create -n qiime2-2017.7 --file https://data.qiime2.org/distro/core/qiime2-2017.7-conda-linux-64.txt
# 激活工作环境
source activate qiime2-2017.7
# 建立工作目录
mkdir -p qiime2-importing-tutorial
cd qiime2-importing-tutorial
导入带质量值的测序数据
地球微生物组标准混样单端数据 “EMP protocol” multiplexed single-end fastq
此类数据标准包括两个文件,扩展名均为fastq.gz,一个是barcode文件,一个是样品混样测序文件。
# 建样品目录
mkdir -p emp-single-end-sequences
# 下载 barcode文件
wget -O "emp-single-end-sequences/barcodes.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/moving-pictures/emp-single-end-sequences/barcodes.fastq.gz"
# 下载序列文件
wget -O "emp-single-end-sequences/sequences.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/moving-pictures/emp-single-end-sequences/sequences.fastq.gz"
# 导入QIIME2格式
qiime tools import \
--type EMPSingleEndSequences \
--input-path emp-single-end-sequences \
--output-path emp-single-end-sequences.qza
地球微生物组标准混样双端数据 “EMP protocol” multiplexed paired-end fastq
此类数据标准包括三个文件,扩展名均为fastq.gz,一个是barcode文件,两个是样品混样测序文件。
# 建样品目录
mkdir -p emp-paired-end-sequences
# 下载序列正向和反向文件
wget -O "emp-paired-end-sequences/forward.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/atacama-soils/1p/forward.fastq.gz"
wget -O "emp-paired-end-sequences/reverse.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/atacama-soils/1p/reverse.fastq.gz"
# 下载barcode文件
wget -O "emp-paired-end-sequences/barcodes.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/atacama-soils/1p/barcodes.fastq.gz"
# 导入QIIME2格式
qiime tools import \
--type EMPPairedEndSequences \
--input-path emp-paired-end-sequences \
--output-path emp-paired-end-sequences.qza
样品文件清单格式 “Fastq manifest” formats
# 下载fastq压缩包zip文件,其中的样品和文件清单文件mainfest
wget -O "se-33.zip" "https://data.qiime2.org/2017.7/tutorials/importing/se-33.zip"
wget -O "se-33-manifest" "https://data.qiime2.org/2017.7/tutorials/importing/se-33-manifest"
wget -O "pe-64.zip" "https://data.qiime2.org/2017.7/tutorials/importing/pe-64.zip"
wget -O "pe-64-manifest" "https://data.qiime2.org/2017.7/tutorials/importing/pe-64-manifest"
# 解压fastq样品文件
unzip -q se-33.zip
unzip -q pe-64.zip
样品清单是包括样品名、文件位置、文件方向三列的csv文件,以pe-64-manifest为例,内容如下:
#样品名、文件位置、文件
sample-id,absolute-filepath,direction
sample1,$PWD/pe-64/s1-phred64-r1.fastq.gz,forward
sample1,$PWD/pe-64/s1-phred64-r2.fastq.gz,reverse
sample2,$PWD/pe-64/s2-phred64-r1.fastq.gz,forward
sample2,$PWD/pe-64/s2-phred64-r2.fastq.gz,reverse
导入质量值不同编码的两类文件Phred33/64 (一般Phred33比较常见,只有非常老的数据才有Phred64格式)
# 导入Phred33格式测序结果
qiime tools import \
--type 'SampleData[SequencesWithQuality]' \
--input-path se-33-manifest \
--output-path single-end-demux.qza \
--source-format SingleEndFastqManifestPhred33
# 导入Phred64格式测序结果
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path pe-64-manifest \
--output-path paired-end-demux.qza \
--source-format PairedEndFastqManifestPhred64
导入OTU表Biom文件
BIOM v1.0.0
# 下载数据并导入为QIIME2的qza格式
wget -O "feature-table-v100.biom" "https://data.qiime2.org/2017.7/tutorials/importing/feature-table-v100.biom"
qiime tools import \
--input-path feature-table-v100.biom \
--type 'FeatureTable[Frequency]' \
--source-format BIOMV100Format \
--output-path feature-table-1.qza
BIOM v2.1.0
wget -O "feature-table-v210.biom" "https://data.qiime2.org/2017.7/tutorials/importing/feature-table-v210.biom"
qiime tools import \
--input-path feature-table-v210.biom \
--type 'FeatureTable[Frequency]' \
--source-format BIOMV210Format \
--output-path feature-table-2.qza
代表性序列 Per-feature unaligned sequence data
wget -O "sequences.fna" "https://data.qiime2.org/2017.7/tutorials/importing/sequences.fna"
qiime tools import \
--input-path sequences.fna \
--output-path sequences.qza \
--type 'FeatureData[Sequence]'
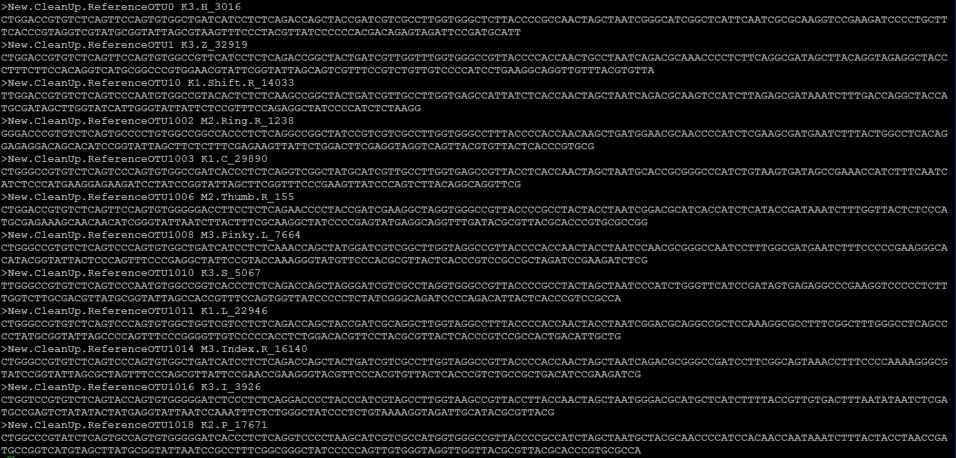
多序列比对后的代表性序列导入(多序列比对后的序列中包括减号,表示比对的gap) Per-feature unaligned sequence data
wget -O "aligned-sequences.fna" "https://data.qiime2.org/2017.7/tutorials/importing/aligned-sequences.fna"
qiime tools import \
--input-path aligned-sequences.fna \
--output-path aligned-sequences.qza \
--type 'FeatureData[AlignedSequence]'
无根进化树导入 Phylogenetic trees (unrooted)
wget -O "unrooted-tree.tre" "https://data.qiime2.org/2017.7/tutorials/importing/unrooted-tree.tre"
qiime tools import \
--input-path unrooted-tree.tre \
--output-path unrooted-tree.qza \
--type 'Phylogeny[Unrooted]'
扩增子分析QIIME2. 6数据导出Exporting data
注:最好按本教程顺序学习,想直接学习本章,至少完成本系列第一篇QIIME2安装。
为什么要导出文件?
QIIME2采用统一qza文件格式,是为了保证文件格式统一和分析流程可追溯。但不可能要求每个人都用此需系统,需要导出其它软件兼容的格式,方便交流和其它用户更个性化的分析。
# 安装QIIME2 2017.7,最少的安装代码,详见QIIME2安装;己安装请跳过
conda update conda
conda create -n qiime2-2017.7 --file https://data.qiime2.org/distro/core/qiime2-2017.7-conda-linux-64.txt
# 激活工作环境
source activate qiime2-2017.7
# 建立工作目录
mkdir -p qiime2-exporting-tutorial
cd qiime2-exporting-tutorial
导出Feature/OTU表
wget -O "feature-table.qza" "https://data.qiime2.org/2017.7/tutorials/exporting/feature-table.qza"
qiime tools export \
feature-table.qza \
--output-dir exported-feature-table
ls -l exported-feature-table/feature-table.biom # 查看结果文件信息
导出的biom文件位于exported-feature-table文件夹中,名为feature-table.biom,可用biom程序对文件进行格式转换和分析
导出进化树
wget -O "unrooted-tree.qza" "https://data.qiime2.org/2017.7/tutorials/exporting/unrooted-tree.qza"
qiime tools export \
unrooted-tree.qza \
--output-dir exported-tree
less exported-tree/tree.nwk
导文件为exported-tree/tree.nwk,是标准树nwk文件

导出与提取
提取包括所有的信息文件,如下例中的feature表文件,结果即包括feature表,又包括生成此文件的相关软件版本信息,还有生成此文件所有步骤的文件说明。
mkdir extracted-feature-table
qiime tools extract \
feature-table.qza \
--output-dir extracted-feature-table
QIIME2. 7元数据 Metadata in QIIME 2
本节分析需要完成1QIIME2安装和2分析实战Moving Picture。
什么是元数据Metadata
元数据是实验设计的描述信息表或统计结果,是分析原始数据必须的基本信息。
元数据是从原始数据中获得生物学发现的关键。在QIIME2中,样品的元数据包括技术细节,如DNA条形码用于区分样品、样品描述,如分类、时间点、取样部分等。对于特征表(Feature,原称OTU)的元数据,一般为特征的注释信息,如物种分类信息。样品和特征表的元数据在QIIME2中很多步分析需要使用。
文本格式实验设计(mapping file)格式要求
为了方便分析,对样品的描述必须包括一些基本信息和格式规范,QIIME2中实验设计mapping file基本要求如下:
- 文件必须是制表符分隔的纯文本文件,建议使用Excel编辑并复制到纯文本编辑器(如editplus, ultraedit等)中保存为txt格式;
- 注释行以#开头,可以出现在文中任意位置,程序会自己忽略;
- 空行也会被忽略;
- 第一行为表头,与QIIME1相比不再以#开头,更合理;
- 表头每列名称必须唯一,不能包括标点符号;(建议实验设计只使用字母和数字,任何符号在后续分析都可能会有问题)
- 文件至少包括除表头外的一行数据;
- 第一列为样品名,用于标识每个样品,必须名字唯一。
下载示例元文件
mkdir qiime2-metadata-tutorial
cd qiime2-metadata-tutorial
# 下载示例样本信息
wget -O "sample-metadata.tsv" "https://data.qiime2.org/2017.7/tutorials/moving-pictures/sample_metadata.tsv"
# 下载失败,尝试下面方法
rm sample-metadata.tsv
wget http://bailab.genetics.ac.cn/markdown/QIIME2/metadata/sample-metadata.tsv
查看示例元文件–实验设计
# 使用less直接查看样品信息
less sample-metadata.tsv
# 使用QIIME2生成网页查看
qiime metadata tabulate \
--m-input-file sample-metadata.tsv \
--o-visualization tabulated-sample-metadata.qzv
# 查看网页结果
qiime tools view tabulated-sample-metadata.qzv
查看结果需要配置ssh方式的可视化,推荐阅读xshell+xmanager或xming配置ssh的X11转发,可图型显示支持;
或下载qzv文件在https://view.qiime2.org/网站在线查看;或直接点击下面链接查看
https://view.qiime2.org/?src=https%3A%2F%2Fdocs.qiime2.org%2F2017.7%2Fdata%2Ftutorials%2Fmetadata%2Ftabulated-sample-metadata.qzv
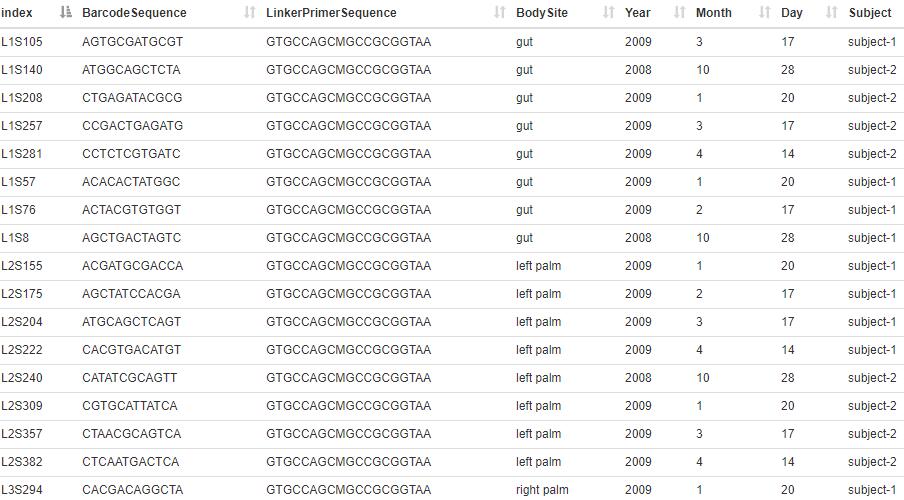
用户思考时间:
上面的示例元文件,subject-1的样品有多少个?来自gut的样品有多少个?
提示:linux下可以使用grep检索;网页中可以按列排序。
查看QIIME2生成的文件元数据
# 下载Alpha多样性PD算法的结果
wget -O "faith_pd_vector.qza" "https://data.qiime2.org/2017.7/tutorials/metadata/faith_pd_vector.qza"
# 结果可视化
qiime metadata tabulate \
--m-input-file faith_pd_vector.qza \
--o-visualization tabulated-faith-pd-metadata.qzv
请最好下载在线查看效果最好,可以网页中直接操作结果(SSH转发的图像操作效果差)?
用户思考时间:
Faith’s PD的最大值是多少?最小值是多少?提示:使用列排序功能。
qzv文件包括结果生成的过程,请查看该结果的追溯图,研究分析过程。
组合型原数据
# 将样品信息和Alpha多样性组合可视化
qiime metadata tabulate \
--m-input-file sample-metadata.tsv \
--m-input-file faith_pd_vector.qza \
--o-visualization tabulated-combined-metadata.qzv
https://view.qiime2.org/?src=https%3A%2F%2Fdocs.qiime2.org%2F2017.7%2Fdata%2Ftutorials%2Fmetadata%2Ftabulated-combined-metadata.qzv 在线查看结果。用户自己的数据 ,需使用qiime tools view命令或下载后在线打开。
查看结果:只包括两个文件中的交集部分,这在以后是很常见的操作,用于选择部分结果。
# 组合Alpha和Beta多样性
wget -O "unweighted_unifrac_pcoa_results.qza" "https://data.qiime2.org/2017.7/tutorials/metadata/unweighted_unifrac_pcoa_results.qza"
qiime emperor plot \
--i-pcoa unweighted_unifrac_pcoa_results.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-file faith_pd_vector.qza \
--o-visualization unweighted-unifrac-emperor-with-alpha.qzv
思才情:那一类取样位置有最高的Faith’s进化多样性值?查看中使用按body site上色,再按Faith’s PD连续着色;
查看特征(Feature/OTU)元数据
wget -O "rep-seqs.qza" "https://data.qiime2.org/2017.7/tutorials/metadata/rep-seqs.qza"
wget -O "taxonomy.qza" "https://data.qiime2.org/2017.7/tutorials/metadata/taxonomy.qza"
qiime metadata tabulate \
--m-input-file rep-seqs.qza \
--m-input-file taxonomy.qza \
--o-visualization tabulated-feature-metadata.qzv
https://view.qiime2.org/?src=https%3A%2F%2Fdocs.qiime2.org%2F2017.7%2Fdata%2Ftutorials%2Fmetadata%2Ftabulated-feature-metadata.qzv 查看代表序列和分类信息的组合结果
用户思考:所有.qza都是元数据吗?使用qiime metadata tabulate自己试试吧。
网页中查看可以导出任意格式的结果,并可追溯实验的分析过程。
扩增子分析QIIME2. 8数据筛选Filtering data
复杂的实验通常会有非常多的组,具体分析中会根据批次、处理条件、基因型等信息进行反复筛选和分析,是分析中常用的操作。本文主讲特征表(Feature/OTU table)和距离矩阵的筛选。
下载实验相关数据
# 激活工作环境
source activate qiime2-2017.7
# 创建工作目录并进入
mkdir qiime2-filtering-tutorial
cd qiime2-filtering-tutorial
# 下载实验设计、特征表和距离矩阵
wget -O "sample-metadata.tsv" "https://data.qiime2.org/2017.7/tutorials/moving-pictures/sample_metadata.tsv"
wget -O "table.qza" "https://data.qiime2.org/2017.7/tutorials/filtering/table.qza"
wget -O "distance-matrix.qza" "https://data.qiime2.org/2017.7/tutorials/filtering/distance-matrix.qza"
# 下载失败的备用链接,成功则跳过
wget http://bailab.genetics.ac.cn/markdown/QIIME2/filter/sample-metadata.tsv
wget http://bailab.genetics.ac.cn/markdown/QIIME2/filter/table.qza
wget http://bailab.genetics.ac.cn/markdown/QIIME2/filter/distance-matrix.qza
过滤Feature/OTU表
按数据量过滤
# 按样品数据量筛选,只保留测序量大于1500的样品(实验中过滤掉数据量过小或过大的样品,有利于发现规律,异常值引起图形严重变形影响分析的判断)
qiime feature-table filter-samples \
--i-table table.qza \
--p-min-frequency 1500 \
--o-filtered-table sample-frequency-filtered-table.qza
# 按Feature的数据量过滤,只有少10个以上reads的Feature/OTU才保留(实验中会有大量低丰度的OTU,它们会增加计算工作量和影响高丰度结果FDR校正pvale的假阴性,通常需要选择一定的阈值进行筛选,常用的有相对丰度千分之五、千分之一、万分之一、万分之一;或者是reads出现的频率至少2、5、10等)
qiime feature-table filter-features \
--i-table table.qza \
--p-min-frequency 10 \
--o-filtered-table feature-frequency-filtered-table.qza
有时也会过滤掉高丰度的OTU或样本,需要使用--p-max-frequency参数
偶然因素的过滤 Contingency-based filtering
举个栗子,你有实验和对照各十组,结果中会有很多OTU只在一个样品中出现,而在其它所有样品中均为零,这种情况一般认为是偶然因素的结果,不具有普遍性,有生物学意义的可能性也比较小,因此通常过滤掉他们,以减少下游分析工作量,降低结果的假阴性率。
# 过滤至少在2个样品中存在的Feature,去除偶然的Feature
qiime feature-table filter-features \
--i-table table.qza \
--p-min-samples 2 \
--o-filtered-table sample-contingency-filtered-table.qza
# 去除小于10个Feature的样品(根据具体情况,有些样品微生物种类极低,可能是异常,如服用过抗生素或PCR扩增出现问题),一般也要筛选后再分析
qiime feature-table filter-samples \
--i-table table.qza \
--p-min-features 10 \
--o-filtered-table feature-contingency-filtered-table.qza
同样上面筛选最小值,有时也会筛选最大值,它们的参数为--p-max-features和--p-max-samples
基于索引的过滤
比如实验中的某些样品发现问题,如生长过程到受胁迫、人或动物吃错药(某些人体样品查出末如实申报的抗生素使用),需要在实验中进行剔除。
# 生成一样需要保留或剔除的样品列表(也可以手动编写文本文件)
echo Index > samples-to-keep.tsv
echo L1S8 >> samples-to-keep.tsv
echo L1S105 >> samples-to-keep.tsv
# 只保留指定的两个样品L1S8和L1S105
qiime feature-table filter-samples \
--i-table table.qza \
--m-metadata-file samples-to-keep.tsv \
--o-filtered-table index-filtered-table.qza
基于实验设计条件的筛选
这是最常用的,重点关注
# 筛选某个条件下一类:实验设计条件Subject列中,名为subject-1的所有样品
qiime feature-table filter-samples \
--i-table table.qza \
--m-metadata-file sample-metadata.tsv \
--p-where "Subject='subject-1'" \
--o-filtered-table subject-1-filtered-table.qza
# 筛选某个条件下多类:身体取样部分中左手和右手的样品
qiime feature-table filter-samples \
--i-table table.qza \
--m-metadata-file sample-metadata.tsv \
--p-where "BodySite IN ('left palm', 'right palm')" \
--o-filtered-table skin-filtered-table.qza
# 同时筛选两个条件共有(和关系/交集):Subject列中subject-1组且在BodySite中的gut
qiime feature-table filter-samples \
--i-table table.qza \
--m-metadata-file sample-metadata.tsv \
--p-where "Subject='subject-1' AND BodySite='gut'" \
--o-filtered-table subject-1-gut-filtered-table.qza
# 同时筛选两个条件均可(或关系/并集):BodySite例为gut或ReportedAntibioticUsage为Yes
qiime feature-table filter-samples \
--i-table table.qza \
--m-metadata-file sample-metadata.tsv \
--p-where "BodySite='gut' OR ReportedAntibioticUsage='Yes'" \
--o-filtered-table gut-abx-positive-filtered-table.qza
# 使用非NOT进行条件筛选:subject-1组中非肠道的样品
qiime feature-table filter-samples \
--i-table table.qza \
--m-metadata-file sample-metadata.tsv \
--p-where "Subject='subject-1' AND NOT BodySite='gut'" \
--o-filtered-table subject-1-non-gut-filtered-table.qza
如果想过滤物种类型呢?需要你的表中有物种信息,尝试使用filter-features选项吧
过滤距离矩阵
# 按样品名过滤
qiime diversity filter-distance-matrix \
--i-distance-matrix distance-matrix.qza \
--m-metadata-file samples-to-keep.tsv \
--o-filtered-distance-matrix index-filtered-distance-matrix.qza
# 按实验设计中的某条件中的组过滤
qiime diversity filter-distance-matrix \
--i-distance-matrix distance-matrix.qza \
--m-metadata-file sample-metadata.tsv \
--p-where "Subject='subject-2'" \
--o-filtered-distance-matrix subject-2-filtered-distance-matrix.qza
扩增子分析QIIME2. 9训练特征分类集Training feature classifiers with q2-feature-classifier
完成此本文分析,必须成功安装QIIME2。
为什么为训练分类集?
因为不同实验的扩增区域不同,鉴定物种分类的精度不同,提前的训练可以让分类结果更准确。
分析前准备
# 安装QIIME2 2017.7,安装过的请跳过
conda update conda
conda create -n qiime2-2017.7 --file https://data.qiime2.org/distro/core/qiime2-2017.7-conda-linux-64.txt
# 激活工作环境
source activate qiime2-2017.7
# 建立工作目录
mkdir training-feature-classifiers
cd training-feature-classifiers
下载和导入参考数据集
# 下载参考OTU数据集
wget -O "85_otus.fasta" "https://data.qiime2.org/2017.7/tutorials/training-feature-classifiers/85_otus.fasta"
# 原文件数据下载缓慢,无法下载尝试我建立的备份
rm 85_otus.fasta
wget http://bailab.genetics.ac.cn/markdown/QIIME2/85_otus.fasta
# 下载参考数据集的物种分类信息
wget -O "85_otu_taxonomy.txt" "https://data.qiime2.org/2017.7/tutorials/training-feature-classifiers/85_otu_taxonomy.txt"
# 下载研究中分析得到的代表性序列文件:示例
wget -O "rep-seqs.qza" "https://data.qiime2.org/2017.7/tutorials/training-feature-classifiers/rep-seqs.qza"
# 导入参考序列
qiime tools import \
--type 'FeatureData[Sequence]' \
--input-path 85_otus.fasta \
--output-path 85_otus.qza
# 导入物种分类信息
qiime tools import \
--type 'FeatureData[Taxonomy]' \
--source-format HeaderlessTSVTaxonomyFormat \
--input-path 85_otu_taxonomy.txt \
--output-path ref-taxonomy.qza
提取参考序列 Extract reference reads
# 按我们测序的引物来提取参考序列中的一段
qiime feature-classifier extract-reads \
--i-sequences 85_otus.qza \
--p-f-primer GTGCCAGCMGCCGCGGTAA \
--p-r-primer GGACTACHVGGGTWTCTAAT \
--p-trunc-len 100 \
--o-reads ref-seqs.qza
训练分类集
# 基于筛选的指定区段,生成实验特异的分类集
qiime feature-classifier fit-classifier-naive-bayes \
--i-reference-reads ref-seqs.qza \
--i-reference-taxonomy ref-taxonomy.qza \
--o-classifier classifier.qza
测试分类集
# 使用训练后的分类集对结果进行注释
qiime feature-classifier classify-sklearn \
--i-classifier classifier.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
# 可视化注释的结果
qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
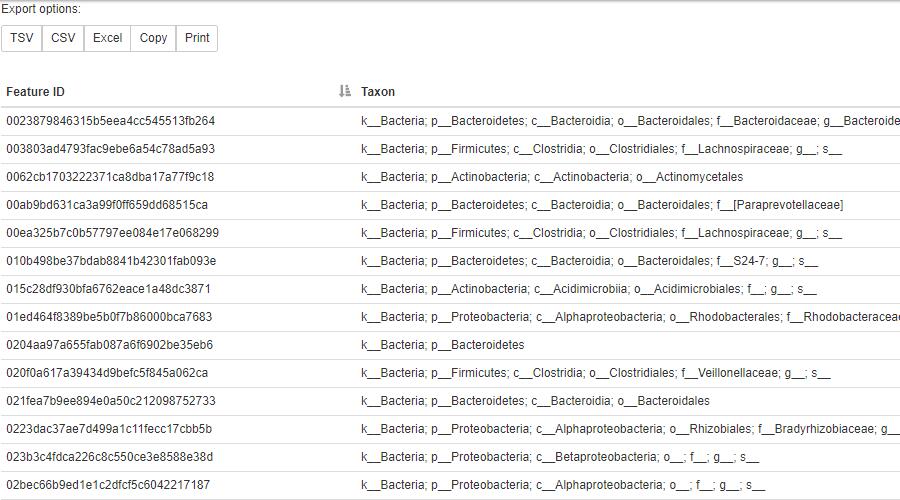
下载并在线view.qiime2.org查看;或点击链接查看预计算的结果https://view.qiime2.org/?src=https%3A%2F%2Fdocs.qiime2.org%2F2017.7%2Fdata%2Ftutorials%2Ffeature-classifier%2Ftaxonomy.qzv
感兴趣的朋友,可以拿这个训练后的结果,和之前的比较。看看有什么变化?
附1. 核心概念
https://docs.qiime2.org/2017.7/concepts/1
想要深入理解QIIME2的分析过程,QIIME定义的基本概念需要了解一下。
- 数据文件: 人工产品 (artifacts)
QIIME2为了使分析流程标准化,分析过程可重复,制定了统一的分析过程文件格式.qza;qza文件类似于一个封闭的系统,里面包括原始数据、分析的过程和结果;这样保证了文件格式的标准,同时可以追溯每一步的分析,以及图表绘制参数。这一方案为实现将来可重复的分析提供了基础。比如文章投稿,同时提供分析过程的文件,别人可以直接学习或重复实验结果。 - 数据文件:可视化(visualizations)
QIIME2生成的图表结果文件类型,以.qzv为扩展名,末尾的v代表visual;它同qza文件类似,包括分析方法和结果,方便追溯图表是如何产生的;唯一与qza不同的,它是分析的终点,即结果的呈现,不会在流程中继续分析。可视化的结果包括统计结果表格、交互式图像、静态图片及其它组合的可视化呈现。这类文件可以使用QIIME2qiime tools view命令查看,不安装程序也可在线 https://view.qiime2.org/78 导入显示; - 语义类型(Semantic types)
QIIME2每步分析中产生的qza文件,都有相应的语义类型,以便程序识别和分析,也避免用户引入不合理的分析过程(如使用末标准化的OTU表进行多样性分析)。了解分析各步的结果,才能对分析有更深入和全面的认识。 - 插件(Plugins)
QIIME2中的某个特定功能即为插件,比如拆分样品、Alpha多样性分析等。插件每个人都可以开发,系列已经由社区开发了标准化分析的插件,其他用户按其标准开发的特定分析,并可与团队联系发布,或整合入平台。 - 方法和可视化
方法是对QIIME2定义的输入格式进行操作的过程,并产生标准格式的输出,以方便后续分析,输入和输出均为qza文件;可视化是对定义的标准输入,产生统计表格或可视化图形,方便用户解读,输入为qza格式,输出为qzv,文件不仅包括结果,还包括处理的分析命令和参数,方便重复和检查分析过程是否准确。
附2. Glossary 名词解释
Action 方法或可视化的动作
A general term for a method or visualizer.
Artifact 本流程定义的文件格式,存储数据和分析结果
Data that can be used as input to a QIIME method or visualizer, or that can be generated as output from a QIIME method. Artifacts typically have the extension .qza when written to file.
Method 对Artifact分析的方法
An action that takes some combination of artifacts and parameters as input, and produces one or more artifacts as output. These output artifacts could subsequently be used as input to other QIIME 2 methods or visualizers. Methods can produce intermediate or terminal outputs in a QIIME analysis.
Parameter 参数,软件或方法中可调整的部分
A primitive (i.e., non-artifact) input to an action. For example, strings, integers, and booleans are primitives. Primitives are never output from an action.
Pipeline 流程,一系统分析方法的串联
A combination of actions. This is not yet implemented.
Plugin 插件,可扩展的功能
A plugin provides microbiome (i.e. domain-specific) analysis functionality that is accessible to users through a variety of interfaces built around the QIIME 2 framework. Plugins can be developed and distributed by anyone. In more technical terms, a plugin is a Python 3 package that instantiates a qiime2.plugin.Plugin object, and registers actions, data formats, and/or semantic types that become discoverable in the QIIME 2 framework.
Result 分析结果
A general term for an artifact or visualization. A result is produced by a method, visualizer, or pipeline.
Visualization 可视化,把数据绘制成图表方便查看和分析规律
Data that can be generated as output from a QIIME visualizer. Visualizations typically have the extension .qzv when written to file.
Visualizer 可视化工具,将结果可视化的软件
An action that takes some combination of artifacts and parameters as input, and produces exactly one visualization as output. Output visualizations, by definition, cannot be used as input to other QIIME 2 methods or visualizers. Visualizers can only produce terminal output in a QIIME analysis.
附3. 常用的语义类型semantic types
原文链接:https://docs.qiime2.org/2017.7/semantic-types/1
FeatureTable[Frequency]: 频率,即Feature表(OTU表),为每个样品中对应OTU出现频率的表格
FeatureTable[RelativeFrequency]: 相对频率,OTU表标准化为百分比的相度丰度
FeatureTable[PresenceAbsence]: OTU有无表,显示样本中某个OTU有或无的表格
FeatureTable[Composition]: 组成表,每个样品中OTU的频率
Phylogeny[Rooted]: 有根进化树
Phylogeny[Unrooted]: 无根进化树
DistanceMatrix: 距离矩阵
PCoAResults: 主成分分析结果
SampleData[AlphaDiversity]: Alpha多样性结果,来自样本自身的分析
SampleData[SequencesWithQuality]: 带质量的序列,要求有质量值,要求序列名称与样品存在对应关系,如为按样品拆分后的数据格式
SampleData[PairedEndSequencesWithQuality]: 成对的带质量序列,要求序列ID与样品编号存在对应关系;
FeatureData[Taxonomy]: 每一个OTU/Feature的分类学信息
FeatureData[Sequence]: 代表性序列
FeatureData[AlignedSequence]: 代表性序列进行多序列比对的结果
FeatureData[PairedEndSequence]: 双端序列进行聚类或去噪后,分类好的OTU或Feature
EMPSingleEndSequences: 采用地球微生物组计划标准实验方法产生的单端测序数据;
EMPPairedEndSequences: 采用地球微生物组计划标准实验方法产生的双端测序数据;
TaxonomicClassifier: 用于物种注释的分类软件
Reference
- https://qiime2.org/4
- Caporaso, J. Gregory, Justin Kuczynski, Jesse Stombaugh, Kyle Bittinger, Frederic D. Bushman, Elizabeth K. Costello, Noah Fierer et al. “QIIME allows analysis of high-throughput community sequencing data.” Nature methods 7, no. 5 (2010): 335-336.
- Schloss, Patrick D., Sarah L. Westcott, Thomas Ryabin, Justine R. Hall, Martin Hartmann, Emily B. Hollister, Ryan A. Lesniewski et al. “Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities.” Applied and environmental microbiology 75, no. 23 (2009): 7537-7541.
- https://docs.qiime2.org/2017.7/glossary/2
- https://docs.qiime2.org/2017.7/install/virtual/docker/
- https://docs.qiime2.org/2017.7/concepts/1
- https://docs.qiime2.org/2017.7/tutorials/moving-pictures/
- Microb Ecol Health Dis. 2015 May 29;26:27663. doi: 10.3402/mehd.v26.27663. eCollection 2015. https://www.ncbi.nlm.nih.gov/pubmed/26028277
- Nat Methods. 2016 Jul;13(7):581-3. doi: 10.1038/nmeth.3869. Epub 2016 May 23. DADA2: High-resolution sample inference from Illumina amplicon data. https://www.ncbi.nlm.nih.gov/pubmed/27214047
- Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns http://msystems.asm.org/content/2/2/e00191-16
- Python交互可视化图形库 http://bokeh.pydata.org/en/latest/1
- https://docs.qiime2.org/2017.7/tutorials/fmt/7
- The data in this tutorial was initially presented in: Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Dae-Wook Kang, James B. Adams, Ann C. Gregory, Thomas Borody, Lauren Chittick, Alessio Fasano, Alexander Khoruts, Elizabeth Geis, Juan Maldonado, Sharon McDonough-Means, Elena L. Pollard, Simon Roux, Michael J. Sadowsky, Karen Schwarzberg Lipson, Matthew B. Sullivan, J. Gregory Caporaso and Rosa Krajmalnik-Brown. Microbiome (2017) 5:10. DOI: 10.1186/s40168-016-0225-7.
- https://docs.qiime2.org/2017.7/tutorials/atacama-soils/3
- The data used in this tutorial is presented in: Arid Soil Microbiome: Significant Impacts of Increasing Aridity. Neilson, Califf, Cardona, Copeland, van Treuren, Josephson, Knight, Gilbert, Quade, Caporaso, and Maier. mSystems (under review).
1F
docker安装