首先来了解一下三个概念:
1、adapter是一段短的序列已知的核酸链,用于链接序列未知的目标测序片段。
2、barcode,也称为index,是一段很短的寡居核酸链,用于在多个样品混合测序时,标记不同的样品。
3、insert是用于测序的目标片段,因为是包括在两个adapter之间,所以被称为“插入”片段。
一个常见测序片段类似与adapter--barcode--insert--adapter。测序开始时前几个碱基无法测得,第一个adapter在数据输出时被去除;由于测序仪读长限制,第二个adapter通常无法测得。所以,经常得到类似 barcode--部分insert的read。最后,把barcode去除,只保留测度insert的片段,这个操作的术语是demultiplexing。但是有时候测序时会测穿,也就是说会得到barcode--insert的read--部分adapter,那么这里就包含了接头了,这里的接头也就是大家经常说去接头要去除的部分。
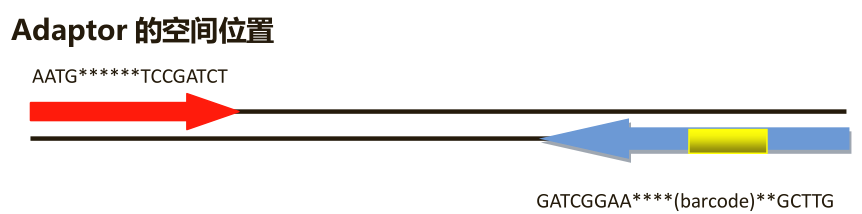
有了以上思路之后,如何去除接头应该就心里有数了,不要照抄网上的教程,没有理解原理有些教程用到你根本不行,虽然有时候你可能按照网上教程不报错并且会看到去除了些,但是这才是最坑的,没去除干净你自己也发觉不了,这还不是最坏的,毕竟接头含量大多数时候较少影响不大,但是如果你做组装或者人品不好的时候,结果就会很出乎意料。
扯这么多,是因为踩过网上教程的坑,现在回归正题
假设我有一个双端测序的数据,最原始的没有去除接头的,然后公司也告诉了我接头序列
接头序列:
- adapt1:AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT
- _REV:AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT
- adapt2:GATCGGAAGAGCACACGTCTGAACTCCAGTCACNNNNNNATCTCGTATGCCGTCTTCTGCTTG
- NNNNNN:barcode
barcode:

以上咱们已经了解了这个数据的基本情况,下一步咱们使用cutadapt进行去除接头操作
跟进cutadapt教程主要有几个参数:
-a:左端reads的接头
-A:右端reads的接头,注意右端出现的接头是因为侧穿了,所以他的接头序列是左端reads的接头的反向互补序列
-m:表示去除接头后如果read长度小于这个m值就不要了
--pair-filter:采用双末端模式来去除接头,保持两端数据匹配
完整命令如下:
- cutadapt -a adapt2 -A adapt1_REV -m 20 --pair-filter=both -o out_fq1 -p out_fq2 fq1 fq2
例子完整命令:
先安装cutadapt:
pip install cutadapt
按照上面流程进行构建命令:
- cutadapt -a GATCGGAAGAGCACACGTCTGAACTCCAGTCACAGTCAAATCTCGTATGCCGTCTTCTGCTTG -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT -m 50 --pair-filter=both -o Sample1_combined_rmAD_R1.fastq -p Sample1_combined_rmAD_R2.fastq Sample1_combined_R1.fastq Sample1_combined_R2.fastq
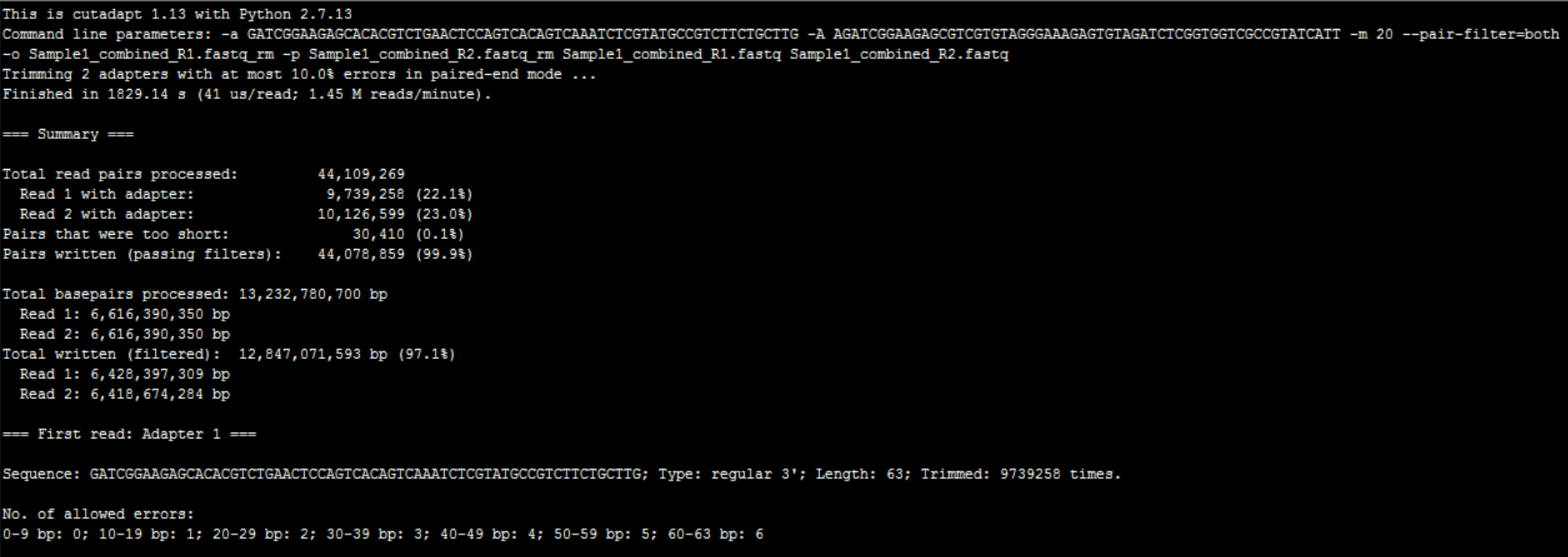
结果如下: