

做为一个合格的生物信息菜鸟,没钱测序咋整,免不了到处求数据呀找数据.....练好基本功必须先从找数据开始!今天小编就来介绍一下一个存储大量高通量数据的的数据库-SRA。
1、简介
SRA(Sequence ReadArchive)数据库是用于存储二代测序的原始数据,包括 454,Illumina,SOLiD,IonTorrent,Helicos 和 CompleteGenomics。除了原始序列数据外,SRA现在也存在raw reads在参考基因的比对信息。
根据SRA数据产生的特点,将SRA数据分为四类:
- Studies-- 研究课题
- Experiments-- 实验设计
- Runs-- 测序结果集
- Samples-- 样品信息
SRA中数据结构的层次关系为:Studies->Experiments->Samples->Runs.
- Studies是就实验目标而言的,一个study 可能包含多个Experiment。
- Experiments包含了Sample、DNA source、测序平台、数据处理等信息。
- 一个Experiment可能包含一个或多个runs。
- Runs 表示测序仪运行所产生的reads。
SRA数据库用不同的前缀加以区分:
- ERP或SRP表示Studies;
- SRS 表示 Samples;
- SRX 表示 Experiments;
- SRR 表示 Runs;
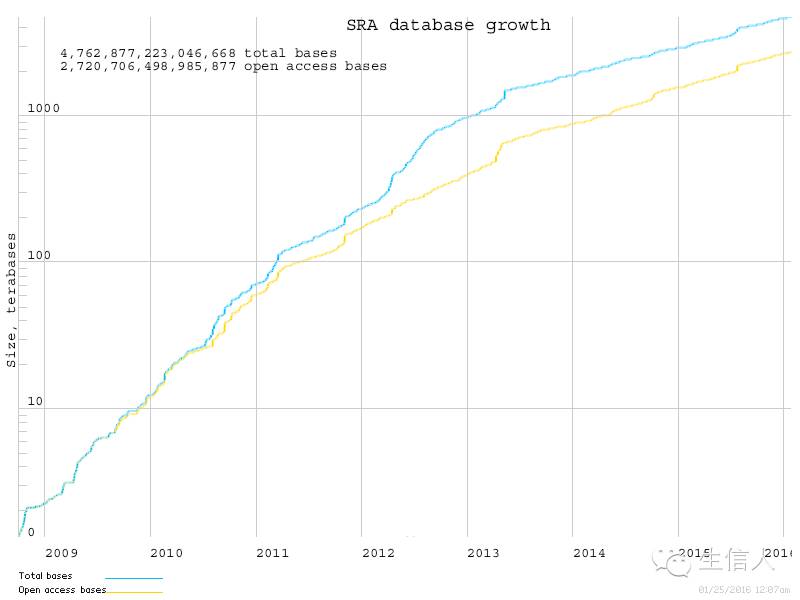
Figure1.SRA数据库收录数据增长曲线
2、使用
 Figure2:搜索相关研究的疾病,选择相应数据集
Figure2:搜索相关研究的疾病,选择相应数据集
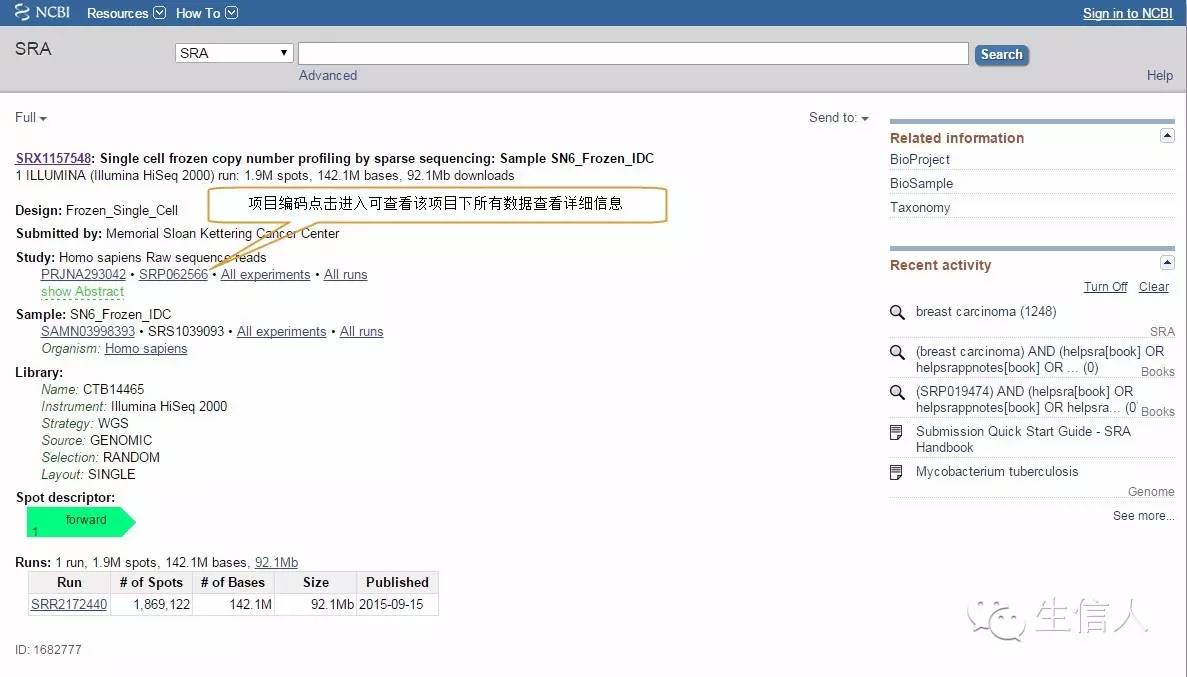 Figure3:点击第一个案例进入详细信息界面
Figure3:点击第一个案例进入详细信息界面
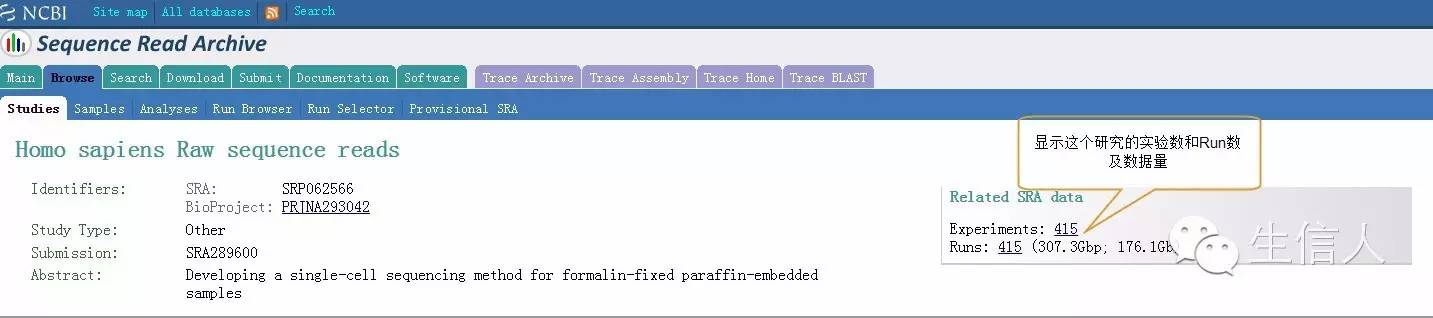
Figure4:Study详细信息页面

Figure4:Experiments详细信息页面

Figure4:Runs详细信息页面,选择要下载的Runs
3、下载数据
要下载SRA数据,我们需要先安装SRA Toolkit软件包,下载地址:
https://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software
根据自己的环境下载相应的软件包。
主要包括:
- CentOS 32/64
- Ubuntu 32/64
- MacOS 32/64
- MS Windows 32/64
以CentOS为例:
1、下载安装:
wget "http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-centos_linux64.tar.gz" tar xzf sratoolkit.current-centos_linux64.tar.gz
2、运行下载
cd sratoolkit.2.5.7-centos_linux64/bin ./prefetch SRR2172038
下载完成后,会在你的工作主目录下生成一个ncbi的文件夹。
cd ncbi/public/sra
查看SRR2172038.sra数据
3、转换fastq
/sratoolkit.2.5.7-centos_linux64/bin/fastq-dump ./SRR2172038.sra
4、转换fasta
/sratoolkit.2.5.7-centos_linux64/bin/fastq-dump --fasta ./SRR2172038.sra
4、数据提交
一个 SRA study 所包含的内容, 应该在一个 LSBI 的项目中提交。 即 SRA study 和 LSBI项目为 1 对 1 关系。 一个 study 的内容可以在一个项目下, 分成几个批次提交, 每次提交不同的内容。
一个批次的 SRA 数据, 包括一个.info 文件和一个名为 DATA, 装有提交原始文件的子文件夹。 子文件夹中内容为描述 metadata 的 xml 文件或者 sff 等格式的数据文件。 一个完整的 study, 包括一个或多个 study.xml, experiment.xml, sample.xml 和 run.xml, 以及一个或多个数据文件。 但是一个批次的提交数据不一定包括所有的文件。
Run.xml 和该 xml 中包括的所有数据文件, 必须要在一个批次中提交。
- 请先确认您已是数据中心网站注册的用户, 否则请登陆中心网站,注册。
- 登陆中心网站后,点击左侧菜单的 mydata,选择已有项目或创建新项目。
- 选择已有批次或创建新批次。 在创建批次时, 选择要提交的数据类型为“SRA”。
- 在点击批次下的 submit data 按钮后, 进入提交页面。
- 首先下载离线提交附件( subdesc.bch), 作为离线提交的标识文件, 是离线提交必须的附件之一。
- 按照 SRA 的数据格式标准, 处理生成数据文件, 连同标识文件一起, 通过提交页面上显示的路径( 为 ftp://shengxin.ren:1221/SRA/****/) 上传至服务器指定目录。
1F
感谢,写的非常详细。