

各种各样的高通量测序和分析平台的运用,使得在一次实验中就能产生数以百计的候选基因,这个时候就需要对其进行注释和筛选。这个过程需要了解每个基因的功能描述和生物学过程以及明确它们是否是G蛋白偶联受体,是否分泌蛋白质,是否在感兴趣的组织中表达等各类问题。虽然每一个注释步骤,都有其对应的专属数据库,比如生物学过程分析数据库GO和通路数据库KEGG等,但是每一个基因如果都在每个数据库中单独走一遍分析流程的话,这显然是不现实的。况且,通常情况下我们会得到一个拥有数十或数百个基因的list,这个时候就需要一个能将各个数据库都整合起来做基因分析的有效工具。
说起基因注释分析工具,很多人第一时间都会想到DAVID。不可否认,DAVID的确能实现基因功能和通路注释等功能。但是!DAVID有一个非常明显的不足,就是数据更新实在是太太太慢了!前一阵大家不是都沉迷于旅行青蛙这种佛系的小游戏嘛,按我说,做生物信息的同胞们不如来看一下DAVID吧,没有比它更佛系的数据资源了,更新频率快慢全凭心情。6.7版本是2010年1月份更新的,然后一直用到了2016年……6.8版本则是2016年5月份更新的,然而现在已经是2018年4月份了,下一个更新版本还遥遥无期……
那么有没有比DAVID更新更快,效果更好的工具呢?答案是:必须滴!今天小编给大家介绍这个秒杀DAVID的工具,叫Metascape。
Metascape的slogan——Fresh、Free & Easy,真的是非常简明扼要地概括了它的核心竞争力。

首先,Metascape的数据每隔月余就会更新一次,最近的一次更新在2018年1月30日。这极大程度保证了数据的时效性和可信度。很幽默的是,在Metascape的官方介绍中,还截图了DAVID的页面,然后将“David died”用红色圈出来,这种正面叫板的操作也是666。
Metascape的数据不仅更新快,其覆盖面也相当广泛。从数据库种类来说,Metascape整合了GO、KEGG、UniProt和DrugBank等多个权威的数据资源,使其不仅能完成通路富集和生物过程注释,还能做基因相关的蛋白质网络分析和涉及到的药物分析,致力于为科研工作者提供每个基因全面而详细的信息。
从所支持的物种数量来说,Metascape不仅能处理人类物种的基因,还能处理包含动物、植物、真菌类和原生生物等多个类别中主要的模式生物的基因。

从数据处理规模上来说,Metascape能一次性处理包含上千个基因的list,并且支持多个不同的gene list同时上传。Metascape对多个不同gene list提供两种分析模式,第一种可以将不同的gene list进行合并分析,从而免去使用者自行合并不同list的烦恼;第二种可以将不同gene list进行独立分析,通过比较找出不同list之间共享或者特异存在的生物过程与通路等。
除了具备以上诸多优点,Metascap被广泛推荐的另一个重要的原因就是它完全不收费,并且使用方式非常非常简单,傻瓜操作级别,出来的结果却非常的酷炫。那么现在就跟着小编开始学习如何应用Metascape吧!
首先,我们来学习Metascape的基础用法——Express Analysis。此用法对非生物信息专业的科研人员堪称亲妈级友好,所有过程只相当于把大象装冰箱——总共分三步,跟着小编走,完全没难度。
第一步:提交基因列表。
打开Metascape的主页(http://metascape.org),页面左侧最明显的一块就是进行Express Analysis的区域
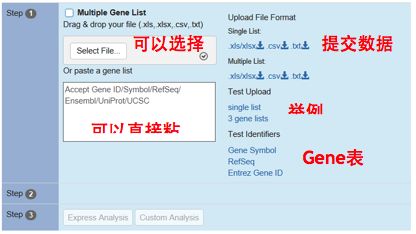
提交基因的时候,可以选择从本地文件上传gene list,如果需要分析的基因数目较少,也可以直接在输入框中粘贴基因。
这一步需要注意的友情小tip:要留心所支持的文件格式与表示基因的方式。Metascape支持3种gene list提交格式,分别是Excel表格,CSV格式与TXT文本格式。在基因列表提交框的右侧,有每一种格式的具体示例可供下载。Metascape支持的基因表示方式也有三种,分别是Gene Symbol,RefSeq ID和Entrez Gene ID,这三种表示方式在输入框的右侧也有具体示例。
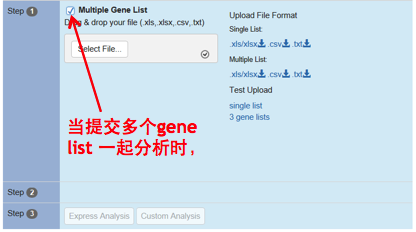
除此之外,当一起提交多个gene list时,还需要特别注意一点:如果想将不同的list分别进行分析,则一定要勾选最上方的【Multiple Gene List】选项,否则Metascape会将多个list整合成一个list一起分析。
为了方便进行后续的结果说明,小编选择了John Dick实验室发布的不同类型的血细胞中的差异表达基因列表,从中抽取了ProB、MLP和MEP三种血细胞的TOP 250差异基因作为测试数据。
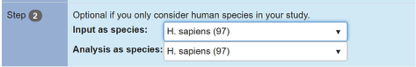
第二步:选择物种信息
选择提交的gene list的物种来源信息和想以哪个物种作为基准来分析。
第三步:点击【Express Analysis】然后开心的等待收结果
Metascape的运行速度不慢。像这次小编提交了3个list共750个gene,也就运行了两三分钟的样子。
Metascape所生成的结果文件无敌酷炫,其中包含很多不同种类的数据和图片。为了方便科研工作者进行使用,Metascape将数据类的结果文件生成Excel统计表格式,供用户进行下载。而图片类的结果,则会被Metascape自动生成一个PPT!!意不意外?!惊不惊喜?!而且这个PPT从布局到配色都做得相当有质感,一股高冷学术风扑面而来~

Metascape还提供所有种类结果文件的打包下载,一个压缩包全部搞定。当然了,作为一个在线应用工具,Metascape也可以在网页上浏览生成的结果报告,但需要注意的是,在线结果报告只能保留72小时,超过72小时之后就需要对数据进行重新分析。
接下来我们以小编的测试数据为例来看看生成的结果吧~
首先是对输入的不同的gene list中的gene进行的数据统计结果:
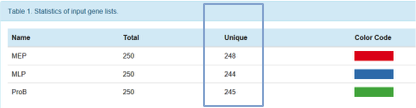
小编每一个list中上传了250个基因,后面unique列的数目表示,该list中,为其独有的gene个数,即这些基因在其他list中不存在。后面的色块表示不同gene list在结果报告中显示的颜色。

这张图也是进一步表示gene list之间的关系。外弧用不同色块表示输入的不同gene list,内圈浅橙色表示这些基因为这个list特有,深橙色表示list之间存在着基因重叠。紫色线表示基因与基因之间的重叠,而蓝色线则表示基因与基因之间存在着功能相关。该图为gene list之间的关联提供了一个宏观的初步印象。
对于输入的每一个gene list,Metascape对其进行通路和生物学过程富集分析时所参考的数据资源有:
KEGG Pathway
GO Biological Processes
生物学反应及信号通路数据库 Reactome Pathway Database
经典通路 Canonical Pathways
哺乳动物蛋白复合物数据库 CORUM
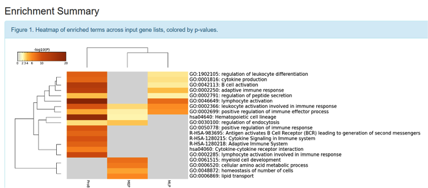
上图即是结果中表示富集到的通路或生物学过程的热图,颜色深浅代表富集程度,后面则是在GO中富集到的term等信息。该图对每一个gene list中的基因主要在那些过程中发挥作用提供了一个初步的阐述。
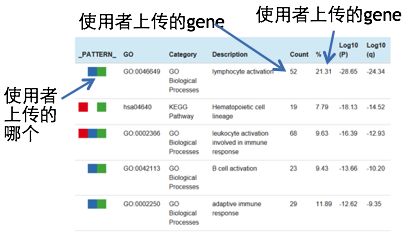
上图会对富集程度比较高的GO term进行单独统计,最左侧一列用色块表示不同的gene list,右侧几列则统计了该list中富集到这个term的gene个数和百分比。以上图的第一行为例,表示MLP与ProB细胞在GO中富集到【淋巴细胞活化】这个term,这与我们所得的先验知识是一致的。
接下来,Metascape会根据每个list富集到的通路和生物学过程,来分群和构建网络。
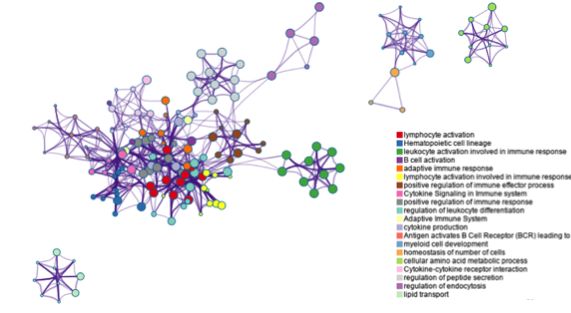
上图是在gene list富集到的term中,挑选出富集程度较高的,然后根据功能相关性聚成几类,并按照关联性和相似性来构建网络。图中用不同的颜色代表不同的类。
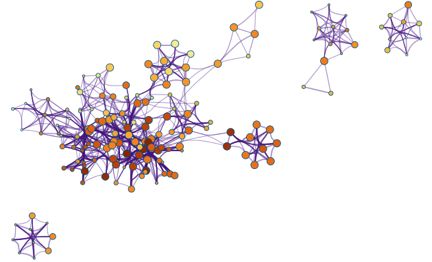
上图则是通过富集程度来着色,颜色越深,表示富集到该类通路或生物学过程的基因数目越多。
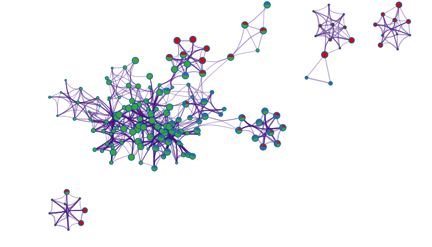
这幅图则是根据不同的gene list来着色。通过以上三幅图之间的比较,用户就可以轻松了解哪个list中的基因以什么程度富集到哪些通路和生物学过程上。值得一提的是,每张网络结果图不仅支持PDF与PNG图片格式下载,还支持以.cys为扩展名的文件格式,使用户可以在下载该文件后直接在Cytoscape上打开,并按照自己的需求对图片进行调整或修改。
Metascape还可以根据基因来生成其相关的蛋白质互作网络。用来构建蛋白质互作网络的数据主要来源于UniProt数据库、BioGRID数据库和inBio Map数据资源等。BioGRID蛋白质互作数据库通过整合多方面经过人工校正或实验验证过的数据集,能提供广泛而全面的蛋白互作信息。而inBio Map数据资源则是由麻省总医院MGH和Broad研究院领衔完成的,提供经过整合的人类蛋白质互作网络。 这些权威的数据资源保证了Metascape结果的可信性。
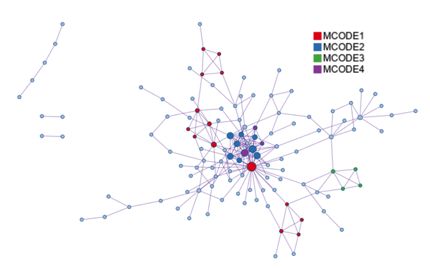
上图以gene list-MEP为例,展现的是MEP基因列表中所有基因相关的蛋白质的全连接互作网络。四种不同的颜色表示互作网络中识别到的模块子结构。
将形成的模块从全连接互作网络中抽象出来,就形成了下图:
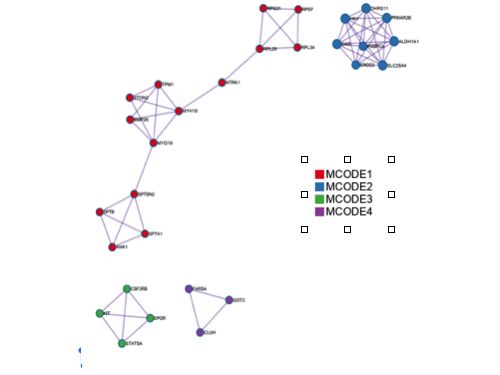
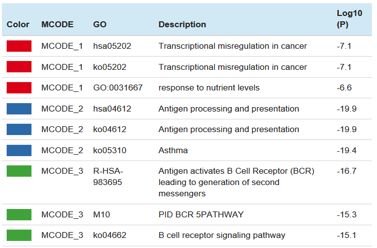
将蛋白质互作数据与通路和生物学过程富集数据相结合,为蛋白质互作网络中识别到的子结构添加生物学意义。每个MOCDE中保留最显著相关的三个GO term,如下图(截图只截取部分)。
每一个gene list所涉及到的蛋白质互作网络都会如上呈现,因篇幅所限,这里就不一一贴图列举了。
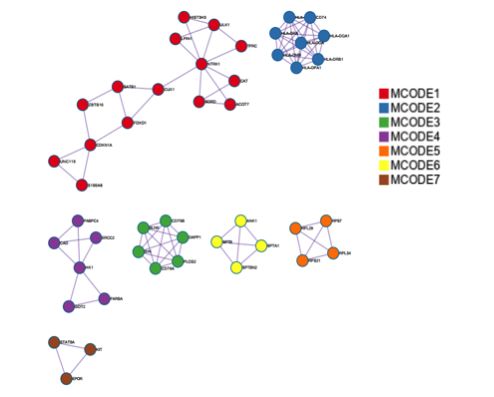
最后Metascape还会将所有的list放在一起,识别蛋白质互作网络中的模块,通过识别到的模块来观察不同list的gene之间是否存在蛋白质互作网络的共享。
下图是从所有list中的gene生成的蛋白质互作网络中抽象出来的子结构图,并以不同颜色来表示不同的模块。
下图用不同的颜色来代表不同list,表示在蛋白质互作模块中,不同list的gene之间的共享关联情况。
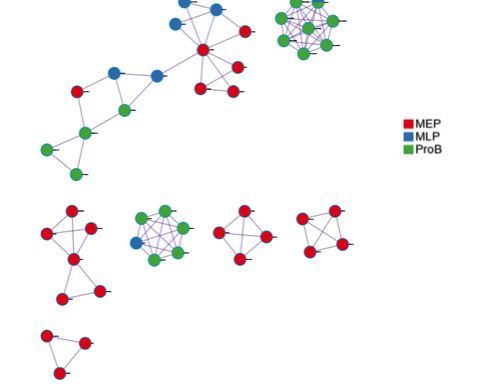
和前面分析单个list时一样,同样在每个MCODE中选取最显著的相关的三个GO term进行列表展示。如下图所示:

以上呢,就是Express Analysis的全部过程和主要的结果介绍。那么除了这种简单友好的一键式操作之外呢,Metascape为拥有一定生物信息学分析基础的用户,提供了更为灵活的进阶用法。即Custom Analysis。
使用时,前两个步骤和Express Analysis是一样的,只有在第三个步骤的时候,换成点击右边的Custom Analysis即可。
![]()
和一般用法不同,进阶用法是一个由四部分组成的workflow。
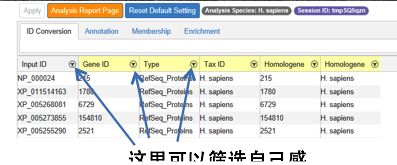
首先,第一个步骤是进行gene ID的转换。虽然在输入时,Metascape支持用户以多种gene ID进行数据输入,但是在实际处理过程中,不论用户输入什么类型的ID,都要先经过转化变成相应的Entrez Gene ID,才能进入后续的分析步骤。在这一步,用户可以根据自己的需求,对提交的基因进行初步筛选。
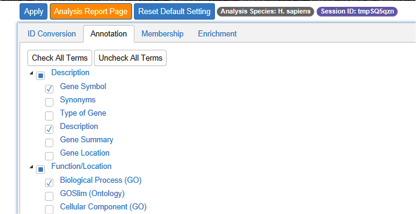
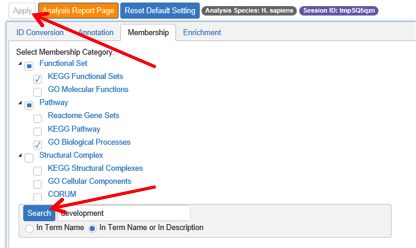
第二个步骤Annotation,用户可以根据自己的需要,选择自己感兴趣的,想在结果中体现的基因注释项目来进行勾选。勾选完成之后,点击左上角的Apply按钮运行。
第三步Membership,支持用户自行选择通路富集、生物过程富集、功能相关和产物分析等每一个注释步骤所用到的数据集,并可以在搜索框中输入感兴趣的字段,比如GO中的某一个或某几个term,或者一些功能性的描述,以便进行更有针对性地分析。
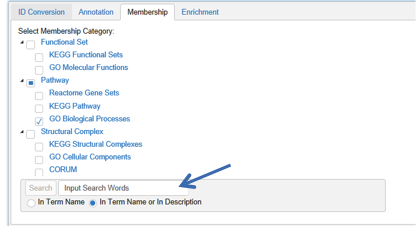
输入完成感兴趣的字段之后,点击左侧的Search按钮进行查找,之后点击左上方Apply生成这一步骤的结果。
最后一个步骤Enrichment,则支持用户选择通路和功能富集过程中的各项指标,以及蛋白质互作网络形成过程中的各项指标。用户可以根据自己的需求,来设定显著性阈值,网络中包含元素的最大或最小值,以及分析步骤中想用到的数据集等参数。
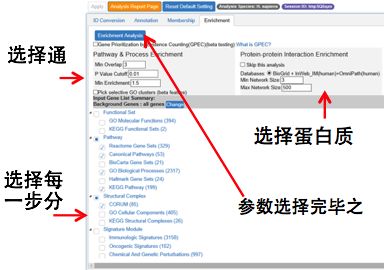
至此,进阶用法的分析步骤就全部完成,生成的结果种类和Express Analysis 所生成的结果种类差不多,这里就不多作介绍了。
1F
你盗用别人公众号的原创文章未免也太无耻了吧 :razz: