methylKit 是一个用于分析甲基化测序数据的R包,不仅支持WGBS,RRBS和目的区域甲基化测序,还支持oxBS-sq, TAB-seq等分析5hmc的数据。 其核心功能是差异甲基化分析和差异甲基化位点和区域的注释。
安装过程如下:
- source(“http://bioconductor.org/biocLite.R“)
- biocLite(“methylKit”)
推荐使用最新版本的R进行安装,这样可以使用最新版本的methylKist。
利用methylKit 做差异分析包括3步
1. 读取原始数据
每个样本一个原始数据,methylKit支持两种格式的methylation calling文件
- 纯文本格式内容如下
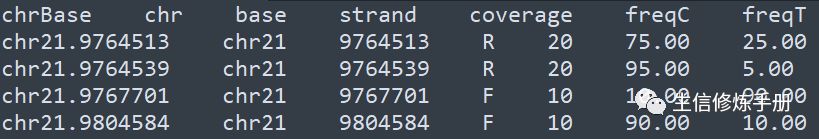
每一行是一个甲基化位点,coverage 代表覆盖这个位点的reads数,freqC 代表甲基化C的比例,freqT 代表非甲基化C的比例。这种纯文本格式内容非常直观,文件大小相比bam 文件小很多,读取的速度更快。
纯文本格式的读取过程如下
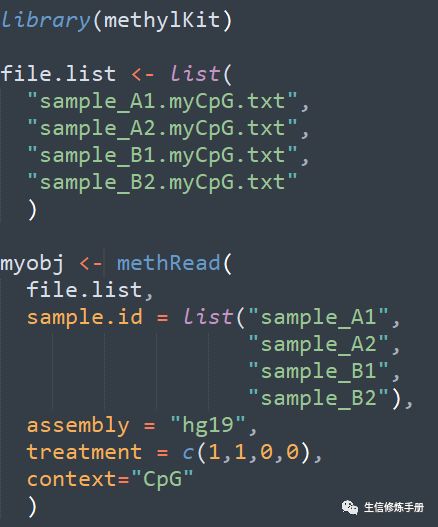
treatment参数指定样本的分组,0代表control组,1代表treatment组
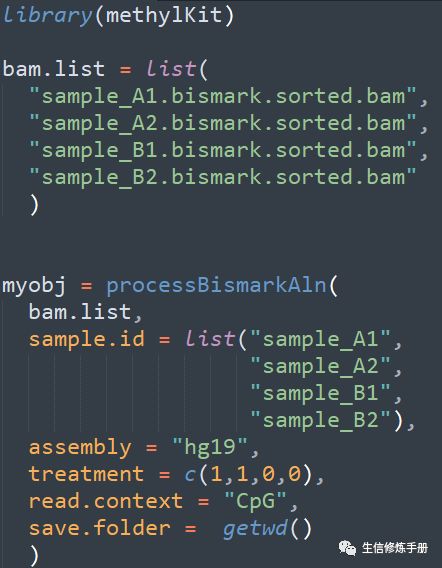
- bam文件
直接读取Bismark软件比对产生的bam文件,通过processBismarkAln实现
用法如下:
2. 合并所有样本的数据
将所有样本的甲基化情况合并,得到所有样本的甲基化表达谱,用法如下
- meth=unite(myobj, destrand=FALSE)
meth 中的内容如下,其实就是之前的methylation calling文件的合并
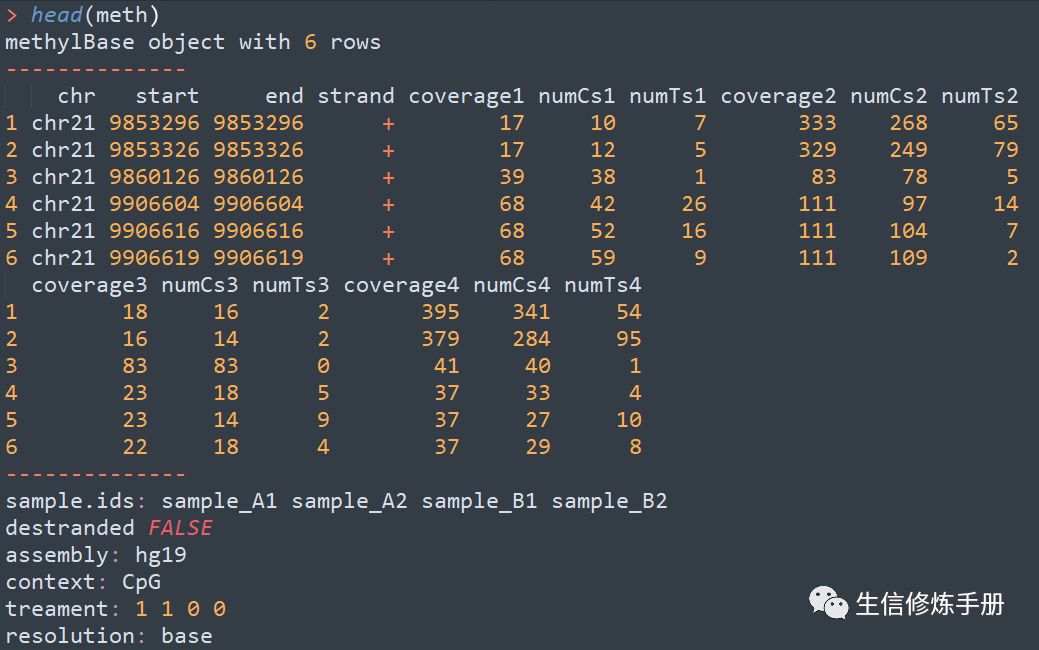
在合并的过程中,默认情况下,只有所有的样本都包含该位点时,才会保留,本质就是取的所有样本的交集,如果你想要取并集,可以修改min.per.group参数的值,该参数的值代表每组中至少有多少个样本覆盖该位点时才保留,如果设置为1,就是取并集。
- meth.min=unite(myobj,min.per.group=1L)
3. 执行差异分析
通过calculateDiffMeth函数来执行差异甲基化分析,用法如下
- myDiff=calculateDiffMeth(meth)
根据甲基化C是变多了还是变少了,可以将差异甲基化的结果分为两大类:
- hypermethylated
- hypomethylated
hypermethylated表示相比control组,treatment组中的甲基化C更多;hypomethylated则相反,表示treatment组中的甲基化C比control组中少。
采用getMethylDiff函数提取差异分析的结果,用法如下
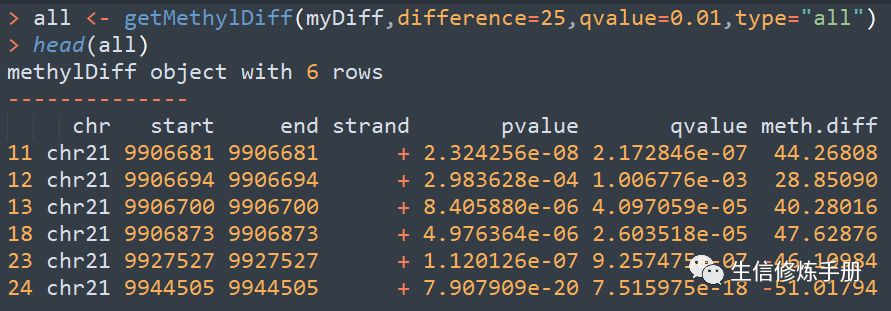
difference函数表明差异的阈值,只有差异大于该阈值时,才会保留,起始就是meth.diff的值,注意是绝对值大于difference的值。
除了difference阈值之外,还有qvalue阈值,小于该阈值的结果保留。在methylKit中,校正p值采用的是SILM算法,和我们常规的BH算法不同。
type参数定义差异的类型,如果你只关注hypermethylated或者hypomethylated,可以设置type 参数的值,单独筛选。
在methylKit中,它的差异分析总是针对合并后的甲基化表达谱,如果你的甲基化表达谱每一行是一个甲基化位点,那么差异分析的结果就是差异甲基化位点;如果你的表达谱每一行是一个甲基化区域,那么差异分析的结果就是差异甲基化区域。上面的例子都是针对差异甲基化位点的,下面看下差异甲基化区域的分析。
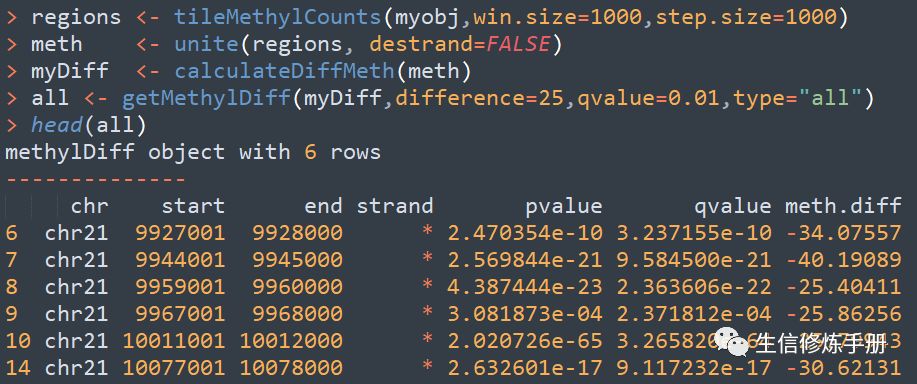
首先遇到的问题就是甲基化区域如何界定,在methylKit中,按照滑动窗口的方式定义甲基化区域,默认窗口大小为10000 bp ,步长为10000bp,通过tileMethylCounts函数实现。
完整的差异甲基化区域分析的代码如下: