

Binning分析指把宏基因组中不同个体微生物序列分开,使得同一类序列聚集在一起的过程,其中常见的是同种菌株的序列聚类在一起。进行binning分析可以宏基因组数据中复杂的功能信息定位到菌株水平,方便后续多组学交叉分析,菌种进化分析等。Binning的原理依据是某一个个体微生物的核酸序列信息(四核苷酸频率、GC含量、必需的单拷贝基因等)和序列丰度(测序覆盖度)是相似的,从而把测序序列聚类成不同的bins。Binning 的算法可分为监督分类和无监督分类,具体可参见 DOI:10.1093/bib/bbs054。
Binning分析可基于三种核酸序列:
- 基于 reads binning:特点是以 reads 为主体,可以聚类出低丰度的菌株信息。因为宏基因组组装过程中对reads的利用率较低,许多 reads 信息在组装后的分析过程中已经丢失。直接基于 reads binning 可以充分利用测序数据。如 Brian Cleary 等 (DOI:10.1038/nbt.3329.Detection) 利用基于 reads binning 的 latent strain analysis 可以聚类出丰度低至0.00001%的菌株。此方法虽然得到更全面的 bins,但低丰度 bins 信息依旧不完整。
- 基于 genes binning:特点是以 genes 为关注主体,适合针对代谢功能的讨论,常用于宏基因组关联分析(MWAS)和多组学联合分析中。因为分析前已经去除冗余的 genes,因此计算资源消耗大大减少。Jun Wang 等 (DOI:10.1038/nrmicro.2016.83) 总结了 MWAS 研究中常见的聚类方法,根据聚类算法和参数的不同,得到的 bins 可以称为:metagenomic linkage groups (MLG)、metagenomic clusters (MGC)、metagenomic species (MGS) 和 metagenomic operational taxonomic units (mOTU)。
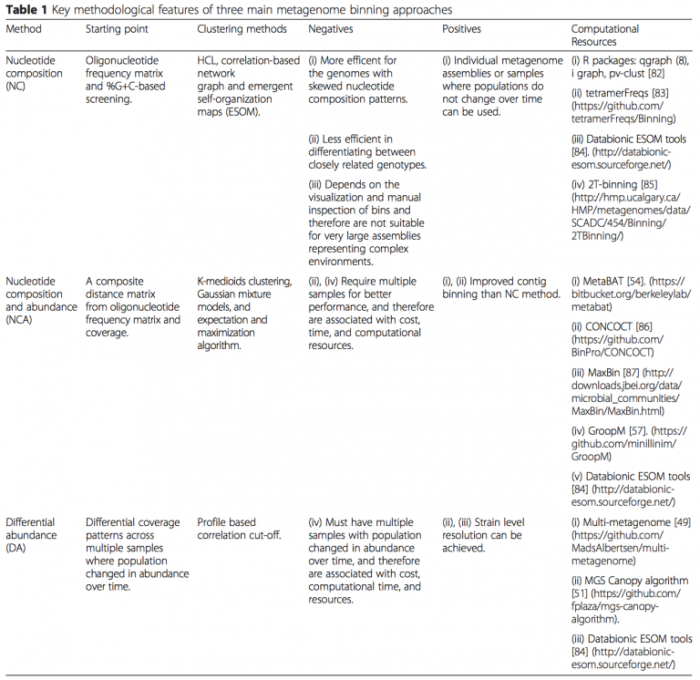
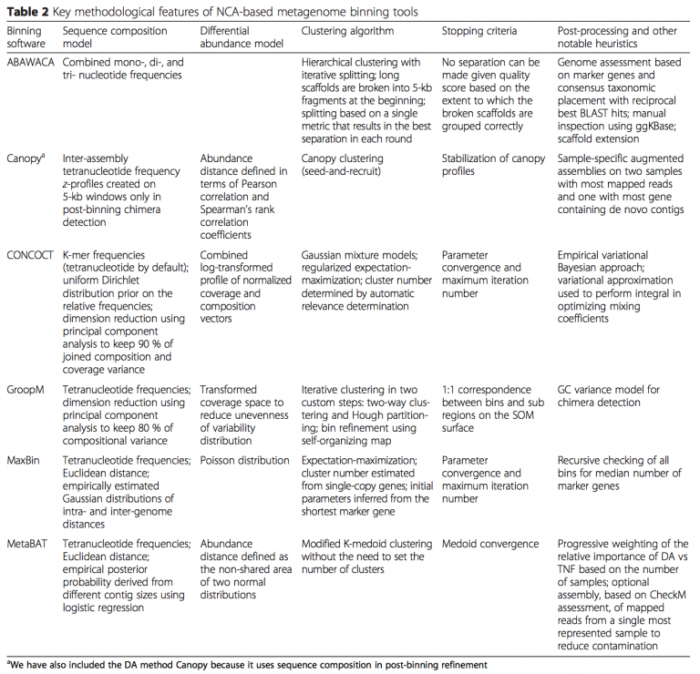
- 基于 contig binning:特点是以 contig 为主体,适合 uncultured 单菌的组装。Naseer Sangwan 等 (DOI: 10.1186/s40168-016-0154-5) 总结了 contig binning 的算法和软件(如下表)。接下来具体介绍其中的 CONCOCT 软件,CONCOCT 自2014年发表在 nature methods 以来,已经被引用超170次。
CONCOCT
1. CONCOCT安装
CONCOCT 提供了两种推荐安装方法:一种基于 Anaconda 安装,另一种基于 Docker 安装。使用 Docker 需要具有管理员权限,不适合普通用户。本文使用 Anaconda 安装。
1.1 关联软件
- ## Fundamental dependencies
- python v2.7.*
- gcc
- gsl
- # Python packages
- cython>=0.19.2
- numpy>=1.7.1
- scipy>=0.12.0
- pandas>=0.11.0
- biopython>=1.62b
- scikit-learn>=0.13.1
- ## Optional dependencies
- # For assembly, use your favorite, here is one
- Velvet
- In velvet installation directory Makefile, set ‘MAXKMERLENGTH=128’, if this value is smaller in the default installation.
- # To create the input table (containing average coverage per sample and contig)
- BEDTools version >= 2.15.0 (only genomeCoverageBed)
- Picard tools version >= 1.110
- samtools version >= 0.1.18
- bowtie2 version >= 2.1.0
- GNU parallel version >= 20130422
- Python packages: pysam>=0.6
- # For validation of clustering using single-copy core genes
- Prodigal >= 2.60
- Python packages: bcbio-gff>=0.4
- R packages: gplots, reshape, ggplot2, ellipse, getopt and grid
- BLAST >= 2.2.28+
1.2 CONCOCT安装
- mkdir /opt/biosoft/concoct
- cd /opt/biosoft/concoct
- # Install Anaconda
- wget https://repo.continuum.io/archive/Anaconda2-4.4.0-Linux-x86_64.sh
- bash Anaconda2-4.4.0-Linux-x86_64.sh
- echo "PATH=/home/train/anaconda2/bin:$PATH" >> ~/.bashrc
- source ~/.bashrc
- # Create a environment of concoct installation
- conda create -n concoct_env python=2.7.6
- source activate concoct_env
- # Install the concoct dependencies
- conda install cython numpy scipy biopython pandas pip scikit-learn
- # Install concoct
- wget https://github.com/BinPro/CONCOCT/archive/0.4.0.tar.gz
- tar zxvf 0.4.0
- cd CONCOCT-0.4.0
- python setup.py install
- echo "PATH=/opt/biosoft/concoct_0.4.1/CONCOCT-0.4.0/scripts:$PATH" >> ~/.bashrc
- source ~/.bashrc
2. CONCOCT运行
CONCOCT的输入数据是组装后的 contig 序列,建议使用 Scaftigs 序列(具体见 宏基因组SOAPdenovo组装),运行前可先去掉长度小于500 bp的短片段序列。
2.1 比对序列至Contig上
通过 bowtie2 将 trimed 序列比对回组装后的 Scaftigs 序列,利用 MarkDuplicates 去除PCR重复。BEDTools 的 genomeCoverageBed 计算 bam 文件中的序列的覆盖率。 CONCOCT 提供了一个sh脚本来完成上述过程(如下)。但可能因为软件安装路径等问题报错,因此接下来我也提供了分步骤命令。
2.1.1 map-bowtie2-markduplicates.sh 运行
- # Usage: bash map-bowtie2-markduplicates.sh [options]
- # -c 启动genomeCoverageBed计算序列覆盖率
- # -t 线程数
- # -k 保留中间文件
- # -p 给bowtie2其他参数. In this case -f
- export MRKDUP=/opt/biosoft/picard-tools-1.77/MarkDuplicates.jar
- for f in $DATA/*.R1.fastq; do
- mkdir -p map/$(basename $f);
- cd map/$(basename $f);
- bash map-bowtie2-markduplicates.sh -ct 1 -p '-f' $f $(echo $f | sed s/R1/R2/) pair $CONCOCT_EXAMPLE/contigs/velvet_71_c10K.fa asm bowtie2;
- cd ../..;
- done
2.1.2 分步运行
- mkdir SampleA
- cd SampleA
- ln -s $DATA/SampleA.fasta ./SampleA.fasta
- # Index reference, Burrows-Wheeler Transform
- bowtie2-build SampleA.fasta SampleA.fasta
- # Align Paired end and bam it
- bowtie2 -p 30 -x SampleA.fasta -1 $Data/SampleA.1.fastq -2 $Data/SampleA.2.fastq -S ./SampleA.sam
- # Index reference for samtools
- samtools faidx SampleA.fasta
- samtools view -bt SampleA.fasta.fai ./SampleA.sam > ./SampleA.bam
- samtools sort -@ 32 ./SampleA.bam -o ./SampleA.sort.bam #如果用1.2版本的samtools,输出不用加bam
- samtools index ./SampleA.sort.bam
- # Mark duplicates and sort
- java -Xms32g -Xmx32g -XX:ParallelGCThreads=15 -XX:MaxPermSize=2g -XX:+CMSClassUnloadingEnabled \
- -jar /opt/biosoft/picard-tools-1.95/MarkDuplicates.jar \
- INPUT=./SampleA.sort.bam \
- OUTPUT=./SampleA.smd.bam \
- METRICS_FILE=./SampleA.smd.metrics \
- AS=TRUE \
- VALIDATION_STRINGENCY=LENIENT \
- MAX_FILE_HANDLES_FOR_READ_ENDS_MAP=1000 \
- REMOVE_DUPLICATES=TRUE
- samtools sort ./SampleA.smd.bam ./SampleA.smds
- samtools index ./SampleA.smds.bam
- samtools flagstat ./SampleA.smds.bam > ./SampleA.smds.flagstat
- # Determine Genome Coverage and mean coverage per contig
- genomeCoverageBed -ibam ./SampleA.smds.bam > ./SampleA.smds.coverage
- awk 'BEGIN {pc=""}
- {
- c=$1;
- if (c == pc) {
- cov=cov+$2*$5;
- } else {
- print pc,cov;
- cov=$2*$5;
- pc=c}
- } END {print pc,cov}' SampleA.smds.coverage | tail -n +2 > SampleA.smds.coverage.percontig
2.2 生成 coverage table
- # usage: gen_input_table.py [-h] [--samplenames SAMPLENAMES] [--isbedfiles] fastafile bamfiles [bamfiles ...]
- # --samplenames 写有样品名的文件,每个文件名一行
- # --isbedfiles 如果在上一步map时运行了genomeCoverageBed,则可以加上此参数后直接用 *smds.coverage文件。如果没运行genomeCoverageBed,则不加此参数,依旧使用bam文件。
- # 若多个样品一起做binning,则样品名都需写入sample.txt;若单个样品,则只写入一个即可
- for i in `ls $DATA/*.1.fastq`
- do
- i=${i/.1.fastq/}
- echo $i
- done > sample.txt
- python gen_input_table.py --isbedfiles --samplenames sample.txt $DATA/SampleA.fasta ./SampleA.smds.coverage > SampleA_concoct_inputtable.tsv
- mkdir ../concoct-input
- mv SampleA_concoct_inputtable.tsv ../concoct-input
2.3 生成 linkage table
- # usage: bam_to_linkage.py [-h] [--samplenames SAMPLENAMES] [--regionlength REGIONLENGTH] [--fullsearch] [-m MAX_N_CORES] [--readlength READLENGTH] [--mincontiglength MINCONTIGLENGTH] fastafile bamfiles [bamfiles ...]
- # --samplenames 写有样品名的文件,每个文件名一行
- # --regionlength contig序列中用于linkage的两端长度 [默认 500]
- # --fullsearch 在全部contig中搜索用于linkage
- # -m 最大线程数,每个ban文件对应一个线程
- # --readlength untrimmed reads长度 [默认 100]
- # --mincontiglength 识别的最小contig长度 [默认 0]
- cd $CONCOCT_EXAMPLE/map
- python bam_to_linkage.py -m 8 --regionlength 500 --fullsearch --samplenames sample.txt $DATA/SampleA.fasta ./SampleA.smds.bam > SampleA_concoct_linkage.tsv
- mv SampleA_concoct_linkage.tsv ../concoct-input
- # 输出文件格式
- # 共2+6*i列 (i样品数),依次为contig1、contig2、nr_links_inward_n、nr_links_outward_n、nr_links_inline_n、nr_links_inward_or_outward_n、read_count_contig1_n、read_count_contig2_n
- # where n represents sample name.
- # Links只输出一次,如 contig1contig2 输出,则 contig2contig1 不输出
- # contig1: Contig linking with contig2
- # contig2: Contig linking with contig1
- # nr_links_inward: Number of pairs confirming an inward orientation of the contigs -><-
- # nr_links_outward: Number of pairs confirming an outward orientation of the contigs <-->
- # nr_links_inline: Number of pairs confirming an outward orientation of the contigs ->->
- # nr_links_inward_or_outward: Number of pairs confirming an inward or outward orientation of the contigs. This can be the case if the contig is very short and the search region on both tips of a contig overlaps or the --fullsearch parameter is used and one of the reads in the pair is outside
- # read_count_contig1/2: Number of reads on contig1 or contig2. With --fullsearch read count over the entire contig is used, otherwise only the number of reads in the tips are counted.
2.4 运行 concoct
- # usage: concoct [-h] [--coverage_file COVERAGE_FILE] [--composition_file COMPOSITION_FILE] [-c CLUSTERS] [-k KMER_LENGTH] [-l LENGTH_THRESHOLD] [-r READ_LENGTH] [--total_percentage_pca TOTAL_PERCENTAGE_PCA] [-b BASENAME] [-s SEED] [-i ITERATIONS] [-e EPSILON] [--no_cov_normalization] [--no_total_coverage] [-o] [-d] [-v]
- # --coverage_file 2.2步骤生成的 coverage table
- # --composition_file contig的fasta格式文件
- # -c, --clusters 最大聚多少类 [默认 400]
- # -k, --kmer_length Kmer长度 [默认 4]
- # -l, -length_threshold 过滤低于多少的contig序列 [默认 1000]
- # -r, --read_length specify read length for coverage, default 100
- # --total_percentage_pca PCA分析中可释方差比例
- # -b, --basename 输出文件路径 [默认当前路径]
- # -s, --seed 聚类的种子数 [0:随机种子,1:默认种子,或其他正整数]
- # -i, --iterations VBGMM的最大迭代次数 [默认 500]
- # -e, --epsilon VBGMM的e-value [默认 1.0e-6]
- # --no_cov_normalization 默认先用对样品做覆盖度均一化,然后对有关的contig做均一化,最后转换日志文件,若设置此参数,则跳过均一化这步
- # --no_total_coverage 默认总的覆盖度会新加一列在覆盖度数据矩阵,若设置此参数则跳过这步
- # -o, --converge_out 输出文件
- cut -f1,3- concoct-input/SampleA_concoct_inputtable.tsv > concoct-input/SampleA_concoct_inputtableR.tsv
- concoct -c 40 --coverage_file concoct-input/SampleA_concoct_inputtableR.tsv --composition_file $DATA/SampleA.fasta -b SampleA_concoct-output/
2.5 concoct 结果可视化
- mkdir evaluation-output
- # 修改ClusterPlot.R中83行的opts(legend.text=theme_text(size=4)为theme(legend.text=element_text(size=4)
- Rscript ClusterPlot.R -c SampleA_concoct-output/clustering_gt1000.csv -p SampleA_concoct-output/PCA_transformed_data_gt1000.csv -m SampleA_concoct-output/pca_means_gt1000.csv -r SampleA_concoct-output/pca_variances_gt1000_dim -l -o evaluation-output/SampleA_ClusterPlot.pdf
结果示例
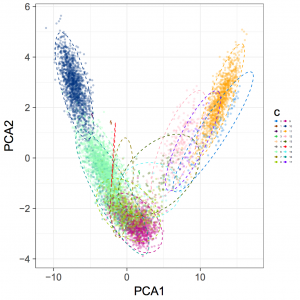
有关binns的完整度和污染评估另起博文介绍。
参考:
WECHAT 生信者言
Mande S S, Mohammed M H, Ghosh T S. Classification of metagenomic sequences: methods and challenges[J]. Briefings in bioinformatics, 2012, 13(6): 669-681.
Sangwan N, Xia F, Gilbert J A. Recovering complete and draft population genomes from metagenome datasets[J]. Microbiome, 2016, 4(1): 8.
Cleary B, Brito I L, Huang K, et al. Detection of low-abundance bacterial strains in metagenomic datasets by eigengenome partitioning[J]. Nature biotechnology, 2015, 33(10): 1053.
Wang J, Jia H. Metagenome-wide association studies: fine-mining the microbiome[J]. Nature Reviews Microbiology, 2016, 14(8): 508-522.