

SOAPdenovo特点
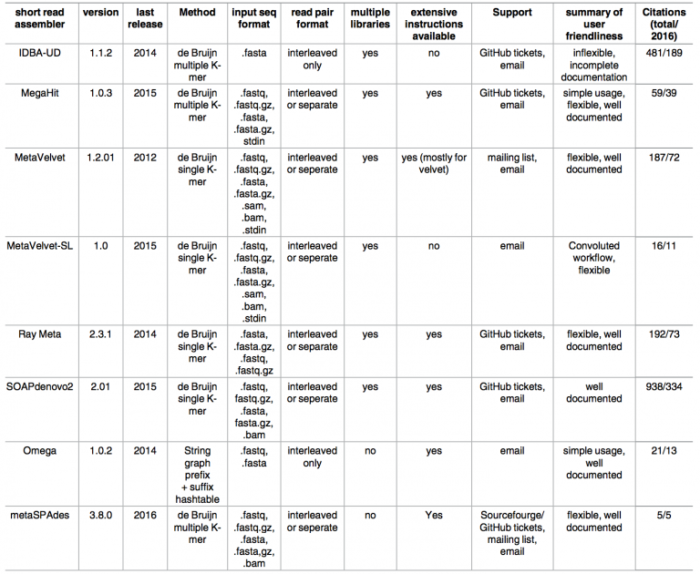
宏基因组denovo组装软件中SOAPdenovo(http://soap.genomics.org.cn/soapdenovo)的使用较为广泛,其具有组装速度快,N50值较好的优点。John Vollmers等人总结了8款宏基因组组装软件的相关信息和特点(DOI:10.1371/journal.pone.0169662),如下表。
SOAPdenovo组装原理
DOI: 10.1101/gr.097261.109
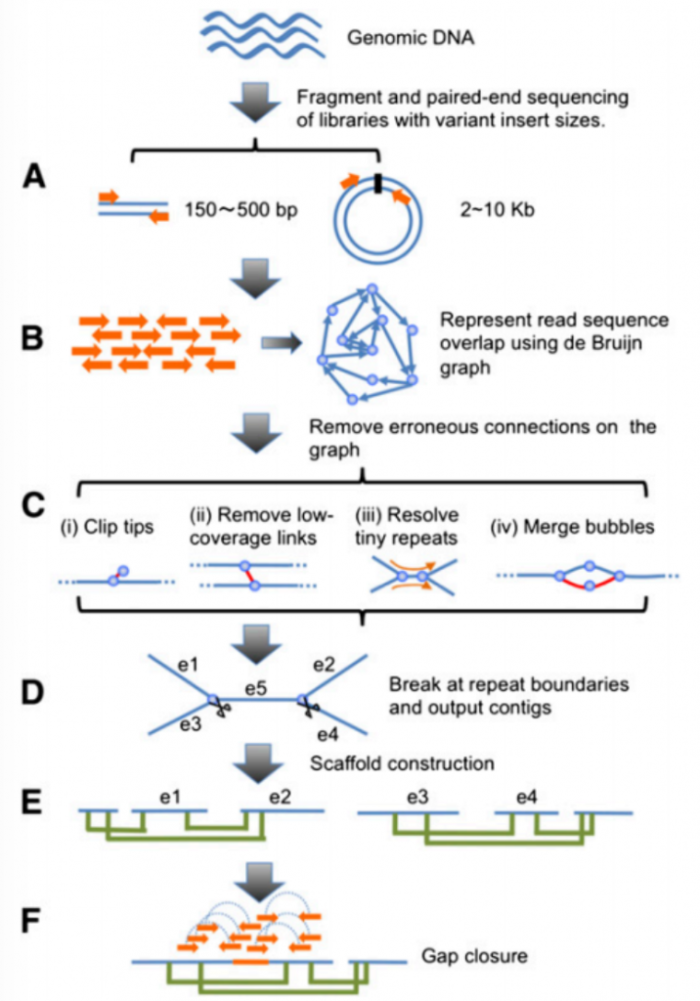
A)基因组总DNA被随机打断,采用双端测序。可考虑建150-500bp的小片段文库和2-10kb的大片段文库;
B)Reads读入后,reads都被切割成某一固定Kmer长度的序列(21bp~127bp),基于Kmer采用de Bruijn数据结构构建Reads间的重叠关系;
C) de Bruijn数据结构图中会产生tips (翼尖)、bubbles (气泡)、low coverage links (低覆盖率链接)、tiny repeat (微小重复)等问题
a.删去tips的短末端
b.删去低覆盖率链接
c.调整read路径解决微小重复
d.删去由重复或者二倍体染色体组的混杂造成bubbles结构;
D)在简化后de Bruijn数据结构图的重复节点打断,输出contigs序列;
E)配对的双端reads比对contigs,基于插入片段长度将单一contigs连成scaffolds;
F)通过配对的双端reads来填补scaffolds中的gaps。
故,SOAPdenovo包含6个工具:pregraph、sparse_pregraph、contig、map、scaffold和all,其中all包括前5个工具。
组装过程
准备测序原始数据。
- mkdir 0.raw_data
- cd 0.raw_data
- gzip -dc ../0.initial_data/A_1.fq.gz > A.1.fastq
- gzip -dc ../0.initial_data/A_2.fq.gz > A.2.fastq
- gzip -dc ../0.initial_data/B_1.fq.gz > B.1.fastq
- gzip -dc ../0.initial_data/B_2.fq.gz > B.2.fastq
- for i in `ls *.1.fastq`
- do
- i=${i/.1.fastq/}
- echo $i
- done > ../samples.txt
- cd ..
NGSQCToolkit
使用NGSQCToolkit(http://www.nipgr.res.in/ngsqctoolkit.html)去除低质量序列得到Clean Data
1) 去除接头序列;
2) 去除所含低质量碱基(质量值≤13)超过40%的 reads;
3) 去除 N 碱基达到5%的 reads。
NGSQCToolkit包括三部分:QC,Triming,Statistics。使用QC中IlluQC.pl去除低质量序列,使用Triming中AmbiguityFiltering.pl去除N碱基较多序列。
- mkdir 1.sequencing_data_quality_control
- cd 1.sequencing_data_quality_control
- # prepare adaptor cluster
- echo "AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT
- CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT
- AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT
- AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT
- CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT
- AGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCGATCTCGTATGCCGTCTTCTGCTTG
- TTTTTTTTTTAATGATACGGCGACCACCGAGATCTACAC
- TTTTTTTTTTCAAGCAGAAGACGGCATACGA
- TACACTCTTTCCCTACACGACGCTCTTCCGATCT
- GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT
- TACACTCTTTCCCTACACGACGCTCTTCCGATCT
- AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTA
- GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT
- AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC" > primer_adaptor.txt
- # remove low quality reads
- for i in `cat ../samples.txt`
- do
- echo "/opt/biosoft/NGSQCToolkit_v2.3.3/QC/IlluQC_PRLL.pl -pe ../0.raw_data/$i.1.fastq ../0.raw_data/$i.2.fastq primer_adaptor.txt 1 -l 60 -s 13 -c 8 -o $i"
- done > command.remove_low_quality_reads.list
- ParaFly -c command.remove_low_quality_reads.list -CPU 4
- # remove non-ATCG reads
- for i in `cat ../samples.txt`
- do
- echo "/opt/biosoft/NGSQCToolkit_v2.3.3/Trimming/AmbiguityFiltering.pl -i ./$i/$i.1.fastq_filtered -irev ./$i/$i.2.fastq_filtered -p 5"
- done > command.remove_non-ATCG_reads.list
- ParaFly -c command.remove_non-ATCG_reads.list -CPU 4
- cd ..
使用SOAPdenovo对样品单独组装
1)对于单个样品,组装时选取 Kmer=55,81,101 等不同值进行组装,比较 N50 的优劣,选取最优结果。通常建议Kmer值为reads长度的55%~85%。
2)将组装得到的 Scaffolds 从 N 连接处打断,得到不含 N 的 Scaftigs序列(continuous sequences within scaffolds);
3)采用 SoapAligner 软件将质控后的 Clean Data 比对至各样品组装后的 Scaftigs 上,获取未被利用上的 reads用于后续混合组装;
4)过滤掉单样品的 Scaftigs 中 500bp 以下的片段用于后续分析。
- mkdir 2.genome_assembling/SOAPdenovo -p
- cd 2.genome_assembling/SOAPdenovo
- for i in `cat ../../samples.txt`
- do
- echo "ln -s ../../1.sequencing_data_quality_control/$i/$i.1.fastq_filtered_trimmed $i.1.fastq"
- echo "ln -s ../../1.sequencing_data_quality_control/$i/$i.2.fastq_filtered_trimmed $i.2.fastq"
- done > command.copy_data.list
- sh command.copy_data.list
- echo "max_rd_len=150
- [LIB]
- avg_ins=300
- reverse_seq=0
- asm_flags=3
- rd_len_cutoff=150
- rank=1
- pair_num_cutoff=3
- map_len=32
- q1=$HOME/2.genome_assembling/SOAPdenovo/A.1.fastq
- q2=$HOME/2.genome_assembling/SOAPdenovo/A.2.fastq" > A_config.txt
- mkdir A
- SOAPdenovo-127mer all -s A_config.txt -o ./A/A -K 81 -d 1 -M 3 -p 8 -R -u -F &> A_soapdenovo.log
- cd A
- fasta_no_blank.pl A.scafSeq > A.scafSeq.noblank && cp A.scafSeq.noblank A.Scaftigs && perl -p -i -e "s/N+/\r\n>\r\n/g" A.Scaftigs
- rename_fasta_identifiers.pl A.Scaftigs > A.Scaftigs.rename
- genome_seq_clear.pl --seq_prefix A_Scaftigs_500 --min_length 500 A.Scaftigs.rename > A_Scaftigs_500.fasta
- cd ..
- #mapping to get unpaired reads
- /opt/biosoft/SOAPaligner/soap2.21release/2bwt-builder A.Scaftigs.rename
- /opt/biosoft/SOAPaligner/soap2.21release/soap -a ../../../2.genome_assembling/SOAPdenovo/A.1.fastq -b ../../../2.genome_assembling/SOAPdenovo/A.2.fastq -D A.Scaftigs.rename.index -o A_PE_output -u A_unmap_output -2 A_SE_output -m 200 -p 20
- echo "max_rd_len=150
- [LIB]
- avg_ins=300
- reverse_seq=0
- asm_flags=3
- rd_len_cutoff=150
- rank=1
- pair_num_cutoff=3
- map_len=32
- q1=$HOME/2.genome_assembling/SOAPdenovo/B.1.fastq
- q2=$HOME/2.genome_assembling/SOAPdenovo/B.2.fastq" > B_config.txt
- mkdir B
- SOAPdenovo-127mer all -s B_config.txt -o ./B/B -K 81 -d 1 -M 3 -p 8 -R -u -F &> B_soapdenovo.log
- cd N
- fasta_no_blank.pl B.scafSeq > B.scafSeq.noblank && cp B.scafSeq.noblank B.Scaftigs && perl -p -i -e "s/N+/\r\n>\r\n/g" B.Scaftigs
- rename_fasta_identifiers.pl B.Scaftigs > B.Scaftigs.rename
- genome_seq_clear.pl --seq_prefix B_Scaftigs_500 --min_length 500 B.Scaftigs.rename > B_Scaftigs_500.fasta
- #mapping to get unpaired reads
- /opt/biosoft/SOAPaligner/soap2.21release/2bwt-builder B.Scaftigs.rename
- /opt/biosoft/SOAPaligner/soap2.21release/soap -a ../../../2.genome_assembling/SOAPdenovo/B.1.fastq -b ../../../2.genome_assembling/SOAPdenovo/B.2.fastq -D B.Scaftigs.rename.index -o B_PE_output -u B_unmap_output -2 B_SE_output -m 200 -p 20
- cd ..
Unmapped Reads混合组装
没有参与组装的测序数据被称为unmapping reads,一般不会参与后续分析。unmapping reads的舍弃直接导致宏基因组数据利用的不完整性。因此,Qin J等提出混合组装。其可以充分利用测序数据,同时有助于挖掘低丰度物种信息。
1)单个样品中未被利用上的reads用于后续混合组装,参数同单个样品组装;
2)将混合组装的 Scaffolds 从 N 连接处打断,得到不含 N 的 Scaftigs 序列;
3)过滤掉混合组装的 Scaftigs 中 500bp 以下的片段用于后续分析。
- mkdir Mix
- cd Mix
- cat $HOME/2.genome_assembling/SOAPdenovo/A/A_unmap_output $HOME/2.genome_assembling/SOAPdenovo/B/B_unmap_output > Mix_seq
- echo "max_rd_len=150
- [LIB]
- avg_ins=300
- reverse_seq=0
- asm_flags=3
- rd_len_cutoff=150
- rank=1
- pair_num_cutoff=3
- map_len=32
- p=$HOME/2.genome_assembling/SOAPdenovo/Mix_seq" > Mix_config.txt
- SOAPdenovo-127mer all -s Mix_config.txt -o ./Mix -K 81 -d 1 -M 3 -p 8 -R -u -F &> Mix_soapdenovo.log
- fasta_no_blank.pl Mix.scafSeq > Mix.scafSeq.noblank && cp Mix.scafSeq.noblank Mix.Scaftigs && perl -p -i -e "s/N+/\r\n>\r\n/g" Mix.Scaftigs
- rename_fasta_identifiers.pl Mix.Scaftigs > Mix.Scaftigs.rename
- genome_seq_clear.pl --seq_prefix Mix_Scaftigs_500 --min_length 500 Mix.Scaftigs.rename > Mix_Scaftigs_500.fasta
参数说明
- -s string 配置文件
- -p int 程序运行时设定的cpu线程数,默认值[8]
- -o string 输出的文件名前缀
- -K int 输入的K-mer值大小,K-mer值必须是奇数,默认值[23]
- -a int 假设初始化内存,为了避免进一步的再分配,默认[0G]
- -d int 为了减少错误测序的影响,去除kmers频数不大于该值(kmerFreqCutoff)的k-mer,默认值[0]
- -R (optional) 利用read鉴别短的重复序列,默认值[NO]
- -D int 为了减少错误测序的影响,去除频数不大于该值的由k-mer连接的边,默认值[1]
- -M int 连contiging时,合并相似序列的强度,最小值0,最大值3,默认值为[1]
- -e int 两边缘之间的弧的权重大于该值,将被线性化,默认值[0]
- -m int 最大的kmer size(max 127)用于multi-kmer,默认[NO]
- -E (optional) 在iterate迭代之前,合并clean bubble功能,仅在当使用multi-kmer时且设置-M参数,默认[NO]
- -F (optional) 利用read对scaffold中的gap进行填补,默认[NO]
- -u (optional) 构建scaffolding前不屏蔽高/低覆盖度的contigs,高频率覆盖度指平均contig覆盖深度的2倍。默认[屏蔽]
- -w (optional) 在scaffold中,保持contigs弱连接于其他contigs,默认[NO]
- -G int 估计gap的大小和实际补gap的大小的差异值,默认值[50bp]
- -L int 用于构建scaffold的contig的最短长度(minContigLen),默认为:[Kmer参数值+2]
- -c float 在scaffolding之前,contigs小于100bp,且覆盖率小于该最小contig覆盖率(c*avgCvg),将被屏蔽,除非参数-u已设定,默认值[0.1]
- -C float 在scaffolding之前,contigs覆盖率大于该最大contigs覆盖率(C*avgCvg),或者contigs小于100bp且覆盖率大于0.8*(C*avgCvg),将被屏蔽,除非参数-u已设定,默认值[2]
- -b float 当处理contigs间的pair-end连接时,如果参数>1,该插入片段的上限值(b*avg_ins)将被用来作为大的插入片段(>1000)的上限,默认值[1.5]
- -B float 如果两个contigs的覆盖率都小于bubble覆盖率(bubbleCoverage)乘以contigs平均覆盖率(bubbleCoverage*avgCvg),则去除在bubble结构中的低覆盖率contig,默认值[0.6]
- -N int 统计基因组大小,默认值[0]
- -V (optional) 输出相关信息(Hawkeye)为了可视化组装,默认[NO]
配置文件
- max_rd_len=150 #read的最大长度
- [LIB] #文库信息以此开头
- avg_ins=300 #文库平均插入长度
- reverse_seq=0 #序列是否需要被反转,插入片段大于等于2k的采用了环化,所以对于插入长度大于等于2k文库,序列需要反转,reverse_seq=1,小片段设为0
- asm_flags=3 #设为1:reads只用于只构建contig;设为2:reads只用于构建scaffold;设为3:reads同时用于构建contig和scaffold;设为4:reads只用于补洞gap
- rank=1 #rank该值取整数,值越低,reads越优先用于构建scaffold
- pair_num_cutoff=3 #该参数规定了连接两个contig或者是pre-scaffold 的可信连接的阈值,即,当连接数大于该值,连接才算有效。小片段文库(<2k)默认值[3],大片段文库默认值[5]。
- map_len=32 #该参数规定了在map过程中reads和contig的比对长度必须达到该值才能作为一个可信的比对。小片段文库一般设32,大片段文库一般设35,默认值[K+2]。
- q1=$DATA_PATH/read_1.fq
- q2=$DATA_PATH/read_2.fq #双端fastq序列,写绝对路径
- f1=./pathfasta_read_1.fa
- f2=./pathfasta_read_2.fa #双端fasta序列,写绝对路径
- q=./pathfastq_read_single.fq #单向fastq
- f=./pathfasta_read_single.fa #单向fasta
- p=./pathpairs_in_one_file.fa #双向测序得到的一个fasta格式的序列文件
输出文件
SOAPdenovo中四部分别对应的输出文件:
pregraph *.kmerFreq *.edge *.preArc *.markOnEdge *.path *.vertex *.preGraphBasic
contig *.contig *.ContigIndex *.updated.edge *.Arc
map *.readOnContig *.peGrads *.readInGap
scaff *.newContigIndex *.links *.scaf *.scaf_gap *.scafSeq *.gapSeq
关键文件如下:
*.contig #没有使用连Scaffold的contig序列
*.scafSeq #SOAPdenovo软件最终的组装序列结果
*.scafStatistics #contigs和scaffolds的最终统计信息
*.Scaftigs #打断N后的Scaftigs序列
*.Scaftigs_500.fasta #长度大于500bp的Scaftigs序列