

做生信的基本上都跟NCBI-SRA打过交道,尤其是fastq-dump大家肯定不陌生.NCBI的fastq-dump软件一直被大家归为目前网上文档做的最差的软件之一",而我用默认参数到现在基本也没有出现过什么问题,感觉好像也没有啥问题, 直到今天看到如下内容, 并且用谷歌搜索的时候,才觉得大家对fastq-dump的评价非常很到位.
![]()
过滤
我们一般使用fastq-dump的方式为
fastq-dump /path/to/xxx.sra
但是这个默认使用方法得到结果往往很糟, 比如说他默认会把双端测序结果保存到一个文件里, 但是如果你加上--split-3之后, 他会把原来双端拆分成两个文件,但是原来单端并不会保存成两个文件. 还有你用--gzip就能输出gz格式, 能够节省空间的同时也不会给后续比对软件造成压力, 比对软件都支持,就是时间要多一点。
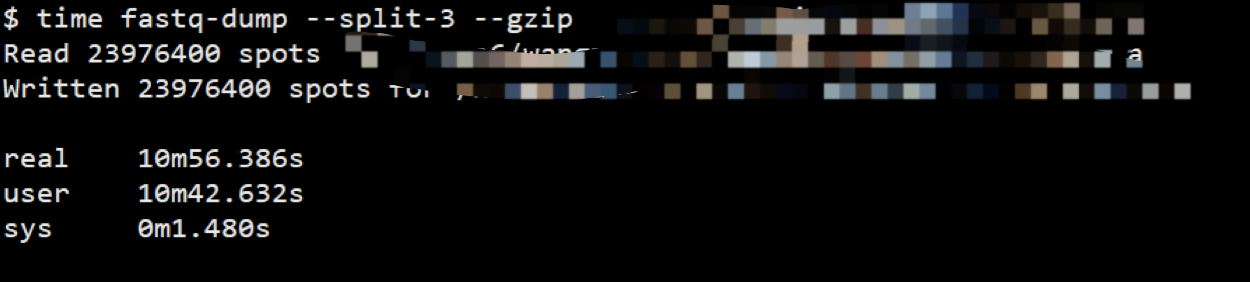
--gzip时间
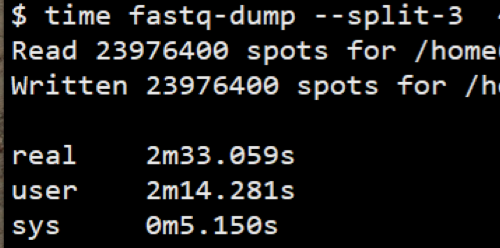
没有gzip时间
但是很不幸运,这些东西在官方文档并没有特别说明,你只有通过不断的踩坑才能学到这些小知识。
我建议尽量去EMBL-EBI去下载原始数据,而不是这种神奇的sra格式,尽管有一些下载的数据其实就是从SRA解压而来。
不过要用fastq-dump,那就介绍几个比较重要的参数吧。我会按照不懂也加,不懂别加,有点意思,没啥意义这三个级别来阐述不同参数的重要级.
太长不看版
如果你不想了解一些细节性内容,用下面的参数就行了
- fastq-dump --gzip --split-3 --defline-qual '+' --defline-seq '@\$ac-\$si/\$ri' SRR_ID
- # 建议加别名
- alias fd='fastq-dump --split-3 --defline-qual '+' --defline-seq '@\\\$ac-\\\$si/\\\$ri' '
数据格式
不懂也加: reads拆分
默认情况下fastq-dump不对reads进行拆分, 对于很早之前的单端测序没有出现问题.但是对于双端测序而言,就会把原本的两条reads合并成一个,后续分析必然会出错.
常见的参数有三类:
--split-spot: 将双端测序分为两份,但是都放在同一个文件中--split-files: 将双端测序分为两份,放在不同的文件,但是对于一方有而一方没有的reads直接丢弃--split-3: 将双端测序分为两份,放在不同的文件,但是对于一方有而一方没有的reads会单独放在一个文件夹里
关于遇到的Rejected 35403447 READS because of filtering out non-biological READS就是因为原来是SE数据,但是用--split-3当作PE数据处理,出现的问题. 看起来好像有问题,但是对后续结果分析没有太多影响.
因此,对于一个你不知道到底是单端还是双端的SRA文件,一律用--split-3.
没事别加: read ID
默认双端测序数据拆分后得到两个文件中同一个reads的名字是一样的,但是加上-I | --readids之后同一个reads的ID就会加上.1和.2进行区分.举个例子
| 是否有-I参数 | ID 1 | ID 2 |
|---|---|---|
| 无 | @SRR5829230.1 1 length=36 | @SRR5829230.1 1 length=36 |
| 有 | @SRR5829230.1.1 1 length=36 | @SRR5829230.1.2 1 length=36 |
问题来了, 明明已经可以通过ID后面的"1"和"2"来区分ID, 加这个参数干嘛. 加完之后还会让后续的BWA报错.所以,没事千万别加
有点意义: 原始格式
默认情况下输出的文件的ID都是SRR开头,但其实原始数据名字不是这样子,比如说@ST-E00600:143:H3LJWALXX:1:1101:5746:1016 2:N:0:CCTCCTGA,@HWI-ST620:248:HB11HADXX:2:1101:1241:2082#0/1这种. 如果你想看到那种格式,而不是SRR,你需要怎么做呢?
可以通过如下三个选项进行修改
F|--origfmt: 仅保留数据名字--defline-seq <fmt>: 定义readsID的显示方式--defline-qual <fmt>: 定义质量的显示方式
其中fmt按照如下要求定义
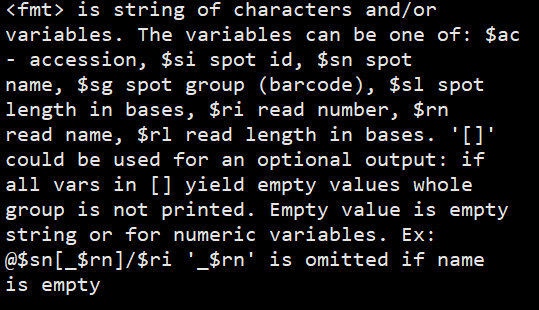
fmt的写法
虽然看起来有点意思,但是对最后的分析其实没啥帮助.
没啥意义: fasta输出
如果下游分析只需要用到fasta文件,那么用--fasta就行. 当然了也有很多方法能够把fastq转换成fasta,比如说samtools.
过滤
我觉得这部分的参数都没有意义, 毕竟完全可以用专门的质控软件处理reads,不过--skip-technical,是唯一比较重要.
- 根据ID:
-N-X - 根据长度:
-M - 多标签序列:
--skip-technical, 这个是唯一有点意思的,就是说如果你原来建库测序使用了多个标签来区分序列, 默认不会输出这个标签. 但是如果不输出标签,我们怎么区分呢? 所以一定要显示声明
有点意思: 输出方式
这部分参数也很重要, 选择是否压缩,还是直接输出到标准输出
--gzip,--bzip2: 压缩方式-Z | --stdout: 输出到标准输出-O|--outdir: 输出到指定文件夹