

ChIP-seq分析过程中,随着测序深度加大,读长数目增多,分析得到的结合位点(‘峰’)的数目也随之增多,但会出现饱和,饱和以后,读长数目增加, ‘峰’的数目基本上不再改变。根据这个规律,为了确定测序得到的读长数目是否足够,可以进行模拟分析在少量测序读长情况下分析计算发现的峰的比例,如下图所示:
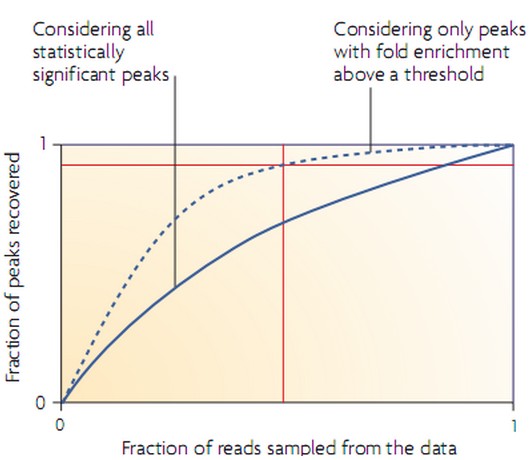
一般情况下,分析得到差异显著的峰的个数随着读长数目的增加而以稳定的比例增加(图中实线所示),这种情况结合位点的数目没有饱和。但是,当对Chip样品和Input DNA样品的峰之间的差异定义一个最小的富集阈值后,分析得到的新的峰的比率逐渐减小(图中虚线所示),这时,当分析足够具有显著差异的结合位点数目的时候,结合位点数目的饱和点出现,可以通过定义几个不同的阈值,分析几个曲线到达平台期的数值来定义饱和的标准(图中桔黄色线所示),所指定的阈值即为最小饱和富集比率(the minimum saturation enrichment ratio,MSER)所得到的最小饱和富集比率可以作为测序深度选择的参数。