

三组不同数据的混合
我们可以从每个数据集(也就是每个批次)中挑选前1000个生物学差异最大的基因
还记得之前是如何得到每个数据集的HVGs吗?主要利用
trendVar、decomposeVar,另外存在多个样本使用combineVar进行合并
整合ID
整合三个数据集的前1000基因后,我们用Reduce()对它们取基因名的交集,最后给基因交集寻找搭配的gene symbol
- rm(list = ls())
- options(stringsAsFactors = F)
- load("gse81076.Rdata")
- load("gse85241.Rdata")
- load("e5601.Rdata")
- # 选择前1000基因
- top.e5601 <- rownames(dec.e5601)[seq_len(1000)]
- top.gse85241 <- rownames(dec.gse85241)[seq_len(1000)]
- top.gse81076 <- rownames(dec.gse81076)[seq_len(1000)]
- # https://www.r-bloggers.com/intersect-for-multiple-vectors-in-r/
- chosen <- Reduce(intersect, list(top.e5601, top.gse85241, top.gse81076))
- # 添加gene symbol
- symb <- mapIds(org.Hs.eg.db, keys=chosen, keytype="ENSEMBL", column="SYMBOL")
- > DataFrame(ID=chosen, Symbol=symb)
- DataFrame with 353 rows and 2 columns
- ID Symbol
- <character> <character>
- 1 ENSG00000115263 GCG
- 2 ENSG00000118271 TTR
- 3 ENSG00000089199 CHGB
- 4 ENSG00000169903 TM4SF4
- 5 ENSG00000166922 SCG5
- ... ... ...
- 349 ENSG00000087086 FTL
- 350 ENSG00000149485 FADS1
- 351 ENSG00000162545 CAMK2N1
- 352 ENSG00000170348 TMED10
- 353 ENSG00000251562 MALAT1
另外,还有一种取交集的方法:先将全部的进行Reduce(),再组合选择前1000
- in.all <- Reduce(intersect, list(rownames(dec.e5601),
- rownames(dec.gse85241), rownames(dec.gse81076)))
- # 设置权重weighted=FALSE ,认为每个batch的贡献一致
- combined <- combineVar(dec.e5601[in.all,], dec.gse85241[in.all,],
- dec.gse81076[in.all,], weighted=FALSE)
- chosen2 <- rownames(combined)[head(order(combined$bio, decreasing=TRUE), 1000)]
取交集的方法会了,但是有个问题不知你有没有注意到:
取交集前提是三个批次之间有相同的HVGs,但是如果对于不同细胞类型的marker基因,它们特异性较强,不一定会出现在所有的batch中
只不过,我们这里只关注交集,因为每个数据集(batch)中的不同donor之间除了marker外,还存在许多表达量又低生物学意义又小的基因,而这些基因用mmCorrect()也不能校正,会给后面的左图带来阻碍,因此这里选择忽略它们
进行基于MNN的校正
简单理解MNN(Mutual nearest neighbors )做了什么
想象一个情况:一个batch(A)中有一个细胞(a),然后再batch(B)中根据所选的feature表达信息找和a最相近的邻居;同样地,对batch B中的一个细胞b,也在batch A中找和它最近的邻居。像a、b细胞这种相互距离(指的是欧氏距离)最近,来自不同batch的作为一对MNN细胞
Haghverdi et al. (2018):MNN pairs represent cells from the same biological state prior to the application of a batch effect

利用MNN pair中细胞间的距离可以用来估计批次效应大小,然后差值可以作为校正批次效应的值
下面就利用mnnCorrect()函数对三个数据集(batch)进行校正批次效应,使用的基因就是chosen 得到的。下面先将三个数据集的表达量信息用logcounts提取出来,并且这个函数做了log的转换,降低了数据的维度;然后将它们放在一个列表中,并根据chosen的基因选择出来前1000个HVGs的表达量信息,是为了后面的循环使用;接着利用do.call() 对每个表达矩阵进行mnnCorrect()操作
- original <- list(logcounts(sce.e5601)[chosen,],
- logcounts(sce.gse81076)[chosen,],
- logcounts(sce.gse85241)[chosen,])
- corrected <- do.call(mnnCorrect, c(original, list(k=20, sigma=0.1)))
- > str(corrected$corrected)
- List of 3
- $ : num [1:353, 1:2285] 0.127 0.137 0.121 0 0.113 ...
- ..- attr(*, "dimnames")=List of 2
- .. ..$ : chr [1:353] "ENSG00000115263" "ENSG00000118271" "ENSG00000089199" "ENSG00000169903" ...
- .. ..$ : chr [1:2285] "HP1502401_J13" "HP1502401_H13" "HP1502401_J14" "HP1502401_B14" ...
- $ : num [1:353, 1:1292] -0.01724 -0.0062 0.01149 0.00689 0.01272 ...
- $ : num [1:353, 1:2346] 0.142 0.138 0.132 0.109 0.11 ...
关于参数的解释:
k表示在定义MNN pair时,设置几个最近的邻居(nearest neighbours ),表示每个batch中每种细胞类型或状态出现的最低频率。增大这个数字,会通过增加MNN pair数量来增加矫正的精度,但是需要允许在不同细胞类型之间形成MNN pair,这一操作又会降低准确性,所以需要权衡这个数字增大k值,会提高precision,降低accuracysigma表示在计算批次效应时如何定义MNN pair之间共享的信息量。较大的值会共享更多信息,就像对同一批次的所有细胞都进行校正;较小的值允许跨细胞类型进行校正,可能会更准确,但会降低精度。默认值为1,比较保守的一个设定,校正不会太多,但多数情况选择小一点的值会更合适减小sigma,会增加accuracy,降低precision这里很有必要说明两个英语词汇:
- Accuracy refers to the closeness of a measured value to a standard or known value. 和标准值比是否"准确"
- Precision refers to the closeness of two or more measurements to each other. 相互之间比是否"精确"
- 另外,提供的original list中各个batch的顺序是很重要的,因为是将第一个batch作为校正的参考坐标系统。一般推荐设置批次效应最大或异质性最强的批次作为对照,可以保证参考批次与其他校正批次之间有充足的MNN pair
检验校正的作用
创建一个新的SingleCellExperiment对象,将三个原始的矩阵和三个校正后的矩阵放在一起
- # omat是原始矩阵,mat是校正后的
- omat <- do.call(cbind, original)
- mat <- do.call(cbind, corrected$corrected)
- # 将mat列名去掉
- colnames(mat) <- NULL
- sce <- SingleCellExperiment(list(original=omat, corrected=mat))
- # 用lapply对三个列表进行循环操作,求列数,为了给rep设置一个重复值
- colData(sce)$Batch <- rep(c("e5601", "gse81076", "gse85241"),
- lapply(corrected$corrected, ncol))
- > sce
- class: SingleCellExperiment
- dim: 353 5923
- metadata(0):
- assays(2): original corrected
- rownames(353): ENSG00000115263 ENSG00000118271 ... ENSG00000170348
- ENSG00000251562
- rowData names(0):
- colnames(5923): HP1502401_J13 HP1502401_H13 ... D30.8_93 D30.8_94
- colData names(1): Batch
- reducedDimNames(0):
- spikeNames(0):
做个t-sne图来看看
图中会显示未校正的细胞是如何根据不同批次分离的,而校正批次后细胞是混在一起的。我们希望这里能够混在一起,是为了后面的分离是真的由于生物差异
- osce <- runTSNE(sce, exprs_values="original", set.seed=100)
- ot <- plotTSNE(osce, colour_by="Batch") + ggtitle("Original")
- csce <- runTSNE(sce, exprs_values="corrected", set.seed=100)
- ct <- plotTSNE(csce, colour_by="Batch") + ggtitle("Corrected")
- multiplot(ot, ct, cols=2)
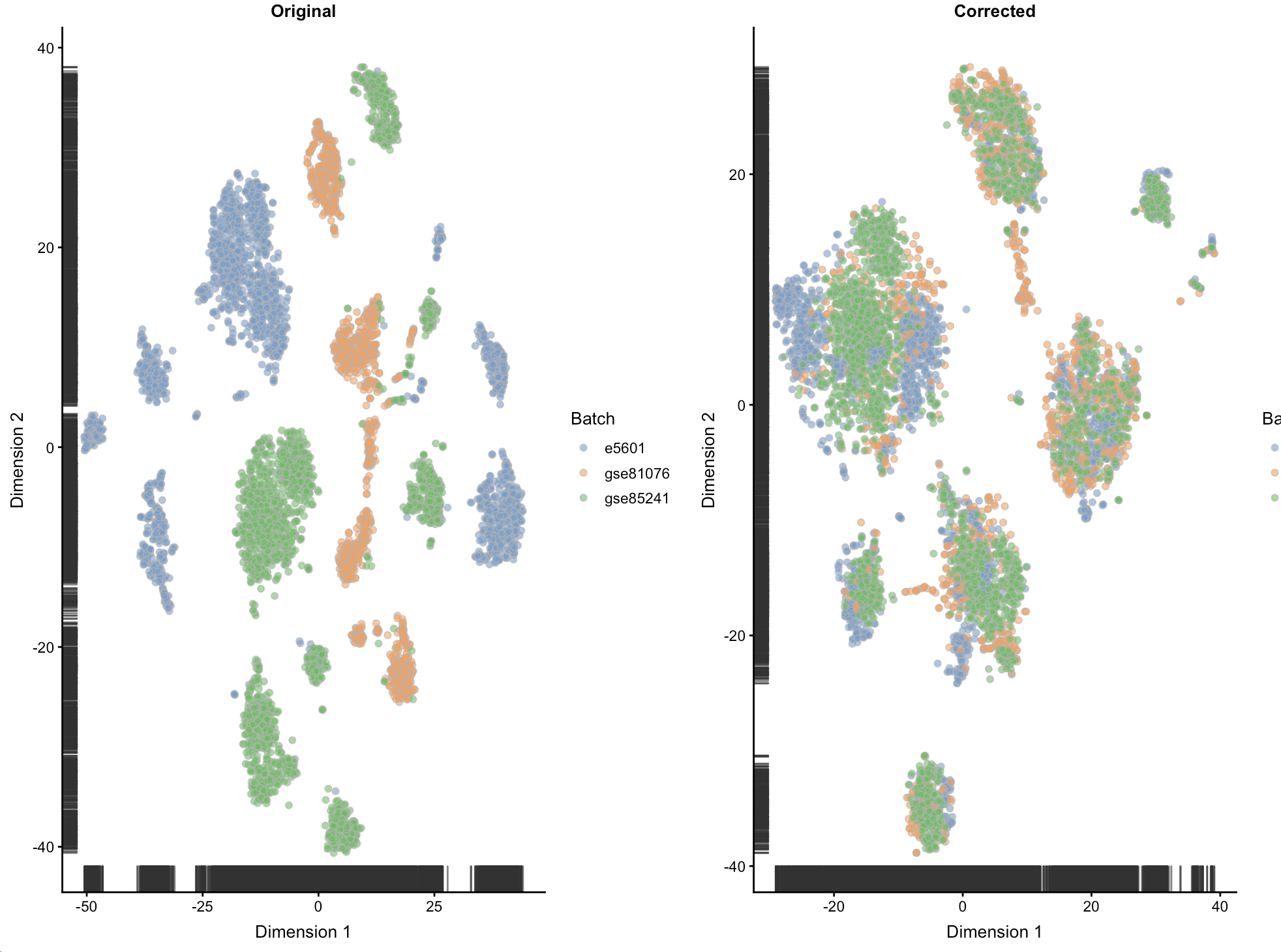
看到E-MTAB-5601这个数据集分离的最严重,推测可能其他数据集采用了UMI
然后再根据几个已知的胰腺细胞的marker基因检测一下,看看这个校正是不是能反映生物学意义。因为如果校正后虽然去除了批次效应,但如果每个群中都体现某个细胞marker基因,对后面分群也是没有意义的。
- ct.gcg <- plotTSNE(csce, by_exprs_values="corrected",
- colour_by="ENSG00000115263") + ggtitle("Alpha cells (GCG)")
- ct.ins <- plotTSNE(csce, by_exprs_values="corrected",
- colour_by="ENSG00000254647") + ggtitle("Beta cells (INS)")
- ct.sst <- plotTSNE(csce, by_exprs_values="corrected",
- colour_by="ENSG00000157005") + ggtitle("Delta cells (SST)")
- ct.ppy <- plotTSNE(csce, by_exprs_values="corrected",
- colour_by="ENSG00000108849") + ggtitle("PP cells (PPY)")
- multiplot(ct.gcg, ct.ins, ct.sst, ct.ppy, cols=2)
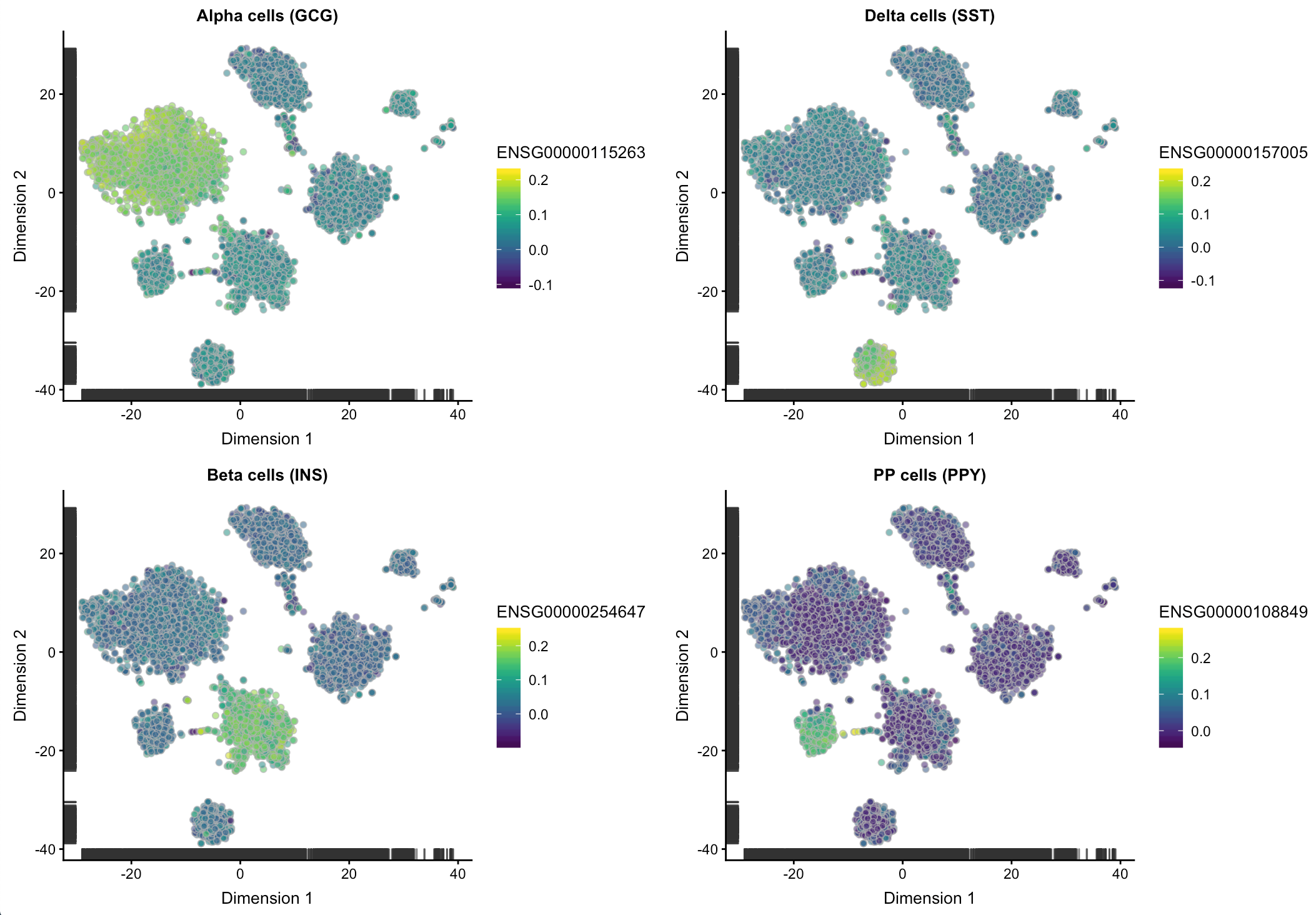
结果可以看到校正后依然可以区分细胞类型,说明既达到了减小批次效应的影响,又能不干扰后续细胞亚型的生物学鉴定-