

第二个数据--CEL-seq2, GSE85241
Muraro et al. (2016) 利用CEL-seq2技术并结合UMI、ERCC得到的
https://www.ncbi.nlm.nih.gov//geo/query/acc.cgi?acc=GSE85241
下面快速使用代码
读数据,看数据
- gse85241.df <- read.table("GSE85241_cellsystems_dataset_4donors_updated.csv.gz", sep='t', header=TRUE, row.names=1)
- > dim(gse85241.df)
- [1] 19140 3072
提取meta信息
- # 还是先看一下
- > head(colnames(gse85241.df))
- [1] "D28.1_1" "D28.1_2" "D28.1_3" "D28.1_4" "D28.1_5" "D28.1_6"
- # 依然是:点号前面的是donor信息
- donor.names <- sub("^(D[0-9]+).*", "\1", colnames(gse85241.df))
- > table(donor.names)
- donor.names
- D28 D29 D30 D31
- 768 768 768 768
- # 然后文章使用了8个96孔板,于是可以将点号和下划线之间的数字提取出来
- plate.id <- sub("^D[0-9]+\.([0-9]+)_.*", "\1", colnames(gse85241.df)) #这句代码中注意使用了一个转义符\,在R中需要用两个反斜线来表示转义
- > table(plate.id)
- plate.id
- 1 2 3 4 5 6 7 8
- 384 384 384 384 384 384 384 384
提取基因、ERCC信息
- gene.symb <- gsub("__chr.*$", "", rownames(gse85241.df))
- is.spike <- grepl("^ERCC-", gene.symb)
- > table(is.spike)
- is.spike
- FALSE TRUE
- 19059 81
基因转换
- library(org.Hs.eg.db)
- gene.ids <- mapIds(org.Hs.eg.db, keys=gene.symb, keytype="SYMBOL", column="ENSEMBL")
- gene.ids[is.spike] <- gene.symb[is.spike]
- keep <- !is.na(gene.ids) & !duplicated(gene.ids)
- gse85241.df <- gse85241.df[keep,]
- rownames(gse85241.df) <- gene.ids[keep]
- > summary(keep)
- Mode FALSE TRUE
- logical 1949 17191
- # 去掉了快2000个重复或无表达的基因
创建单细胞对象
- # 存储metadata作为colData、基因信息作为rawData、ERCC作为spike-in
- sce.gse85241 <- SingleCellExperiment(list(counts=as.matrix(gse85241.df)),
- colData=DataFrame(Donor=donor.names, Plate=plate.id),
- rowData=DataFrame(Symbol=gene.symb[keep]))
- isSpike(sce.gse85241, "ERCC") <- grepl("^ERCC-", rownames(gse85241.df))
质控和标准化
- sce.gse85241 <- calculateQCMetrics(sce.gse85241, compact=TRUE)
- QC <- sce.gse85241$scater_qc
- low.lib <- isOutlier(QC$all$log10_total_counts, type="lower", nmad=3)
- low.genes <- isOutlier(QC$all$log10_total_features_by_counts, type="lower", nmad=3)
- high.spike <- isOutlier(QC$feature_control_ERCC$pct_counts, type="higher", nmad=3)
- data.frame(LowLib=sum(low.lib), LowNgenes=sum(low.genes),
- HighSpike=sum(high.spike, na.rm=TRUE))
- # LowLib LowNgenes HighSpike
- # 577 669 696
- # 然后去掉低质量的细胞
- discard <- low.lib | low.genes | high.spike
- sce.gse85241 <- sce.gse85241[,!discard]
- > summary(discard)
- Mode FALSE TRUE
- logical 2346 726
可以看到文库小的有577个,基因表达少的有669个,高spike-in的有696个,但是最后只去掉了726个,这是因为,有的细胞同时存在以上两种或三种低质量情况,因此并不能简单认为总共去除细胞数=577+669+696
聚类
- clusters <- quickCluster(sce.gse85241, min.mean=0.1, method="igraph")
- > table(clusters)
- clusters
- 1 2 3 4 5 6
- 237 248 285 483 613 480
标准化
- sce.gse85241 <- computeSumFactors(sce.gse85241, min.mean=0.1, clusters=clusters)
- summary(sizeFactors(sce.gse85241))
- sce.gse85241 <- computeSpikeFactors(sce.gse85241, general.use=FALSE)
- summary(sizeFactors(sce.gse85241, "ERCC"))
- sce.gse85241 <- normalize(sce.gse85241)
鉴定HVGs
- block <- paste0(sce.gse85241$Plate, "_", sce.gse85241$Donor)
- fit <- trendVar(sce.gse85241, block=block, parametric=TRUE)
- dec <- decomposeVar(sce.gse85241, fit)
- plot(dec$mean, dec$total, xlab="Mean log-expression",
- ylab="Variance of log-expression", pch=16)
- is.spike <- isSpike(sce.gse85241)
- points(dec$mean[is.spike], dec$total[is.spike], col="red", pch=16)
- curve(fit$trend(x), col="dodgerblue", add=TRUE)
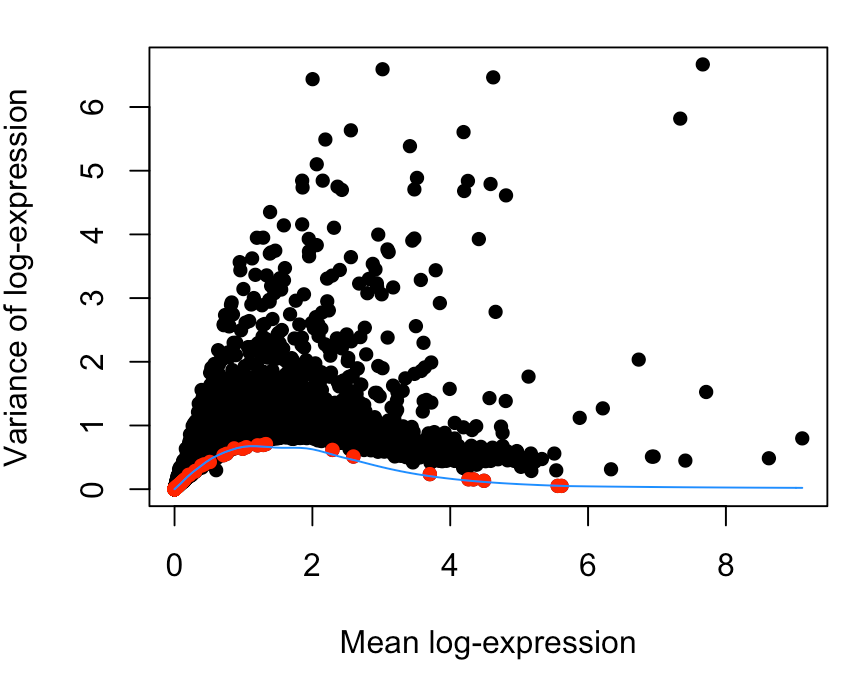
这张图中的ERCC表达量就有一些比较高的,但是占比不高,另外总体波动不大
- # 选出来这些基因
- dec.gse85241 <- dec
- dec.gse85241$Symbol <- rowData(sce.gse85241)$Symbol
- dec.gse85241 <- dec.gse85241[order(dec.gse85241$bio, decreasing=TRUE),]
- > head(dec.gse85241,2)
- DataFrame with 2 rows and 7 columns
- mean total bio
- <numeric> <numeric> <numeric>
- ENSG00000115263 7.66453729345785 6.66863456231166 6.63983282676052
- ENSG00000089199 4.62375793902937 6.46558866721711 6.34422879524839
- tech p.value FDR Symbol
- <numeric> <numeric> <numeric> <character>
- ENSG00000115263 0.0288017355511366 0 0 GCG
- ENSG00000089199 0.12135987196872 0 0 CHGB
第三个数据--Smart-seq2, E-MTAB-5061
Segerstolpe et al. (2016)利用Smart-seq2,添加了ERCC,这个数据和上面两个不同,它存放在ArrayExpress数据库,当然也是用链接规律的:https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5061/ (这个文件比较大,压缩文件151M,解压后700多M)

读入数据
文件较大,先读入样本,也就是第一行(nrow=1),看下数量
- header <- read.table("pancreas_refseq_rpkms_counts_3514sc.txt",
- nrow=1, sep="t", comment.char="")
- # 先看下header信息
- > header[1,1:4]
- V1 V2 V3 V4
- #samples HP1502401_N13 HP1502401_D14 HP1502401_F14
- # 然后将第一个(#samples)去掉
- ncells <- ncol(header) - 1L #保存为整数
然后只加载基因名称和表达矩阵
- # 这段代码需要再好好理解下
- col.types <- vector("list", ncells*2 + 2)
- col.types[1:2] <- "character"
- col.types[2+ncells + seq_len(ncells)] <- "integer"
- e5601.df <- read.table("pancreas_refseq_rpkms_counts_3514sc.txt",
- sep="t", colClasses=col.types)
- # 最后将基因信息和表达矩阵分离
- gene.data <- e5601.df[,1:2]
- e5601.df <- e5601.df[,-(1:2)]
- colnames(e5601.df) <- as.character(header[1,-1])
- dim(e5601.df)
- ## [1] 26271 3514
判断ERCC
- # gene.data[,2]对应测序数据中的基因ID,gene.data[,1]是相应的symbol ID
- is.spike <- grepl("^ERCC-", gene.data[,2])
- > table(is.spike)
- is.spike
- FALSE TRUE
- 26179 92
基因转换
- library(org.Hs.eg.db)
- gene.ids <- mapIds(org.Hs.eg.db, keys=gene.data[,1], keytype="SYMBOL", column="ENSEMBL")
- gene.ids[is.spike] <- gene.data[is.spike,2]
- # 去掉重复和无表达基因
- keep <- !is.na(gene.ids) & !duplicated(gene.ids)
- e5601.df <- e5601.df[keep,]
- rownames(e5601.df) <- gene.ids[keep]
- > summary(keep)
- Mode FALSE TRUE
- logical 3367 22904
提取metadata信息
- metadata <- read.table("E-MTAB-5061.sdrf.txt", header=TRUE,
- sep="t", check.names=FALSE)
- m <- match(colnames(e5601.df), metadata$`Assay Name`)
- stopifnot(all(!is.na(m)))
- metadata <- metadata[m,]
- donor.id <- metadata[["Characteristics[individual]"]]
- > table(donor.id)
- donor.id
- AZ HP1502401 HP1504101T2D HP1504901 HP1506401
- 96 352 383 383 383
- HP1507101 HP1508501T2D HP1509101 HP1525301T2D HP1526901T2D
- 383 383 383 384 384
创建单细胞对象
- sce.e5601 <- SingleCellExperiment(list(counts=as.matrix(e5601.df)),
- colData=DataFrame(Donor=donor.id),
- rowData=DataFrame(Symbol=gene.data[keep,1]))
- isSpike(sce.e5601, "ERCC") <- grepl("^ERCC-", rownames(e5601.df))
后面的操作和之前保持一致了
- sce.e5601 <- calculateQCMetrics(sce.e5601, compact=TRUE)
- QC <- sce.e5601$scater_qc
- low.lib <- isOutlier(QC$all$log10_total_counts, type="lower", nmad=3)
- low.genes <- isOutlier(QC$all$log10_total_features_by_counts, type="lower", nmad=3)
- high.spike <- isOutlier(QC$feature_control_ERCC$pct_counts, type="higher", nmad=3)
- low.spike <- isOutlier(QC$feature_control_ERCC$log10_total_counts, type="lower", nmad=2)
- data.frame(LowLib=sum(low.lib), LowNgenes=sum(low.genes),
- HighSpike=sum(high.spike, na.rm=TRUE), LowSpike=sum(low.spike))
- # LowLib LowNgenes HighSpike LowSpike
- # 162 572 904 359
- # 舍弃低质量细胞
- discard <- low.lib | low.genes | high.spike | low.spike
- sce.e5601 <- sce.e5601[,!discard]
- > summary(discard)
- Mode FALSE TRUE
- logical 2285 1229
- # 聚类
- clusters <- quickCluster(sce.e5601, min.mean=1, method="igraph")
- > table(clusters)
- clusters
- 1 2 3 4 5 6
- 305 307 469 272 494 438
- # 标准化
- sce.e5601 <- computeSumFactors(sce.e5601, min.mean=1, clusters=clusters)
- sce.e5601 <- computeSpikeFactors(sce.e5601, general.use=FALSE)
- sce.e5601 <- normalize(sce.e5601)
因为这个数据中donor信息比较多,所以可视化也要特别对待
- donors <- sort(unique(sce.e5601$Donor))
- > donors
- [1] "AZ" "HP1502401" "HP1504101T2D" "HP1504901"
- [5] "HP1506401" "HP1507101" "HP1508501T2D" "HP1509101"
- [9] "HP1525301T2D" "HP1526901T2D"
一共10个donor,作图可以设置这个参数,调整图片为2列
- par(mfrow=c(ceiling(length(donors)/2), 2),
- mar=c(4.1, 4.1, 2.1, 0.1))
代码作图,注意这段代码和之前的不同
- collected <- list() # 第一行可以先不管,目的是创建一个空列表
- # 下面进行一个循环,对10个donor进行循环:先取出第一个donor的列信息,然后使用if判断它是不是大于两列(也就是说:这个donor是不是有两个以上的细胞样本),如果只有一列那么就舍去;然后对这个donor的所有列进行标准化,去掉细胞文库差异;接着利用trendVar和decomposeVar鉴定HVGs,然后和之前一样进行可视化;最后将这个donor鉴定出来的HVGs信息放入collected这个列表中,留着以后用
- for (x in unique(sce.e5601$Donor)) {
- current <- sce.e5601[,sce.e5601$Donor==x]
- if (ncol(current)<2L) { next }
- current <- normalize(current)
- fit <- trendVar(current, parametric=TRUE)
- dec <- decomposeVar(current, fit)
- plot(dec$mean, dec$total, xlab="Mean log-expression",
- ylab="Variance of log-expression", pch=16, main=x)
- points(fit$mean, fit$var, col="red", pch=16)
- curve(fit$trend(x), col="dodgerblue", add=TRUE)
- collected[[x]] <- dec
- }
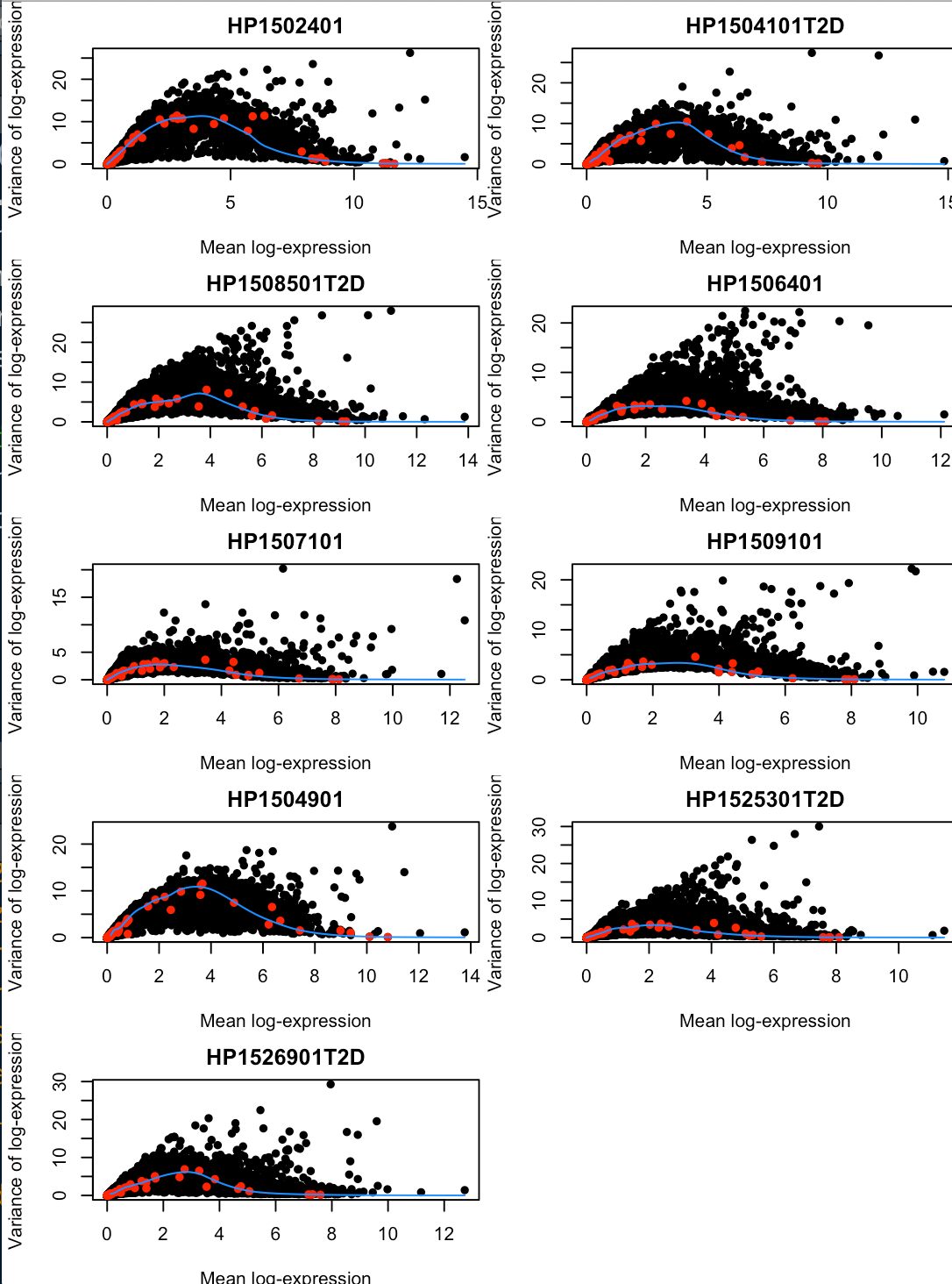
因为这个数据中donor信息比较多,因此我们需要将不同donor的HVGs整合成一个数据框(注意是更高级的SV4数据框)
- dec.e5601 <- do.call(combineVar, collected)
- dec.e5601$Symbol <- rowData(sce.e5601)$Symbol
- dec.e5601 <- dec.e5601[order(dec.e5601$bio, decreasing=TRUE),]
- > head(dec.e5601,3)
- DataFrame with 3 rows and 7 columns
- mean total bio
- <numeric> <numeric> <numeric>
- ENSG00000115263 9.79547495804957 24.9059740209558 24.693297105741
- ENSG00000118271 10.3601718361198 19.0590510324402 18.9670050741979
- ENSG00000089199 8.78499018265489 17.2605488560106 16.9971950283286
- tech p.value FDR Symbol
- <numeric> <numeric> <numeric> <character>
- ENSG00000115263 0.212676915214769 0 0 GCG
- ENSG00000118271 0.0920459582422512 0 0 TTR
- ENSG00000089199 0.263353827682004 0 0 CHGB