

1 前言
依旧是我的传统:不🙅♀️翻译 并结合自己的知识尝试加入自己的理解
这次的官方教程主要介绍使用简单数据集(Lun et al. 2017)来走scRNA的分析流程。这个数据集中包含2个96孔板416B 细胞(一种永生型小鼠骨髓祖细胞系),利用Smart-seq2建库。另外在文库制备过程中加入了ERCC。基因定量是将reads比对到外显子区域,同样每个spike-in转录本也比对到spike-in参考序列(一般会在构建基因组索引前就将ERCC序列加到基因组最后)。当然,这是上游分析的内容,这里我们不关心,实际情况中也并不是人人都要自己做上游分析。我们一般会拿到表达矩阵(这里我们会使用 E-MTAB-5522 这个数据集)
2 关于下载
官方利用了BiocFileCache 包进行下载,我们把下载的数据都存放在自定义的目录中(例如:raw_data ),这样以后再次使用就可以避免重新下载
- BiocManager::install("BiocFileCache")
- library(BiocFileCache)
- # browseVignettes('BiocFileCache') 查看帮助文档
- # 首先自定义一个存放数据的目录
- bfc <- BiocFileCache("raw_data", ask = FALSE)
- # file.path可以是本地路径或者网络链接(直接下载到BiocFileCache指定的目录)
- lun.zip <- bfcrpath(bfc,
- file.path("https://www.ebi.ac.uk/arrayexpress/files",
- "E-MTAB-5522/E-MTAB-5522.processed.1.zip"))
- lun.sdrf <- bfcrpath(bfc,
- file.path("https://www.ebi.ac.uk/arrayexpress/files",
- "E-MTAB-5522/E-MTAB-5522.sdrf.txt"))
- unzip(lun.zip, exdir=tempdir())
3 数据的设置
3.1 加载表达矩阵
前面说到这个数据集包含了两个细胞板测序数据,结果就有两个文件。每个文件的行是内源基因或者spike-in转录本,每列表示细胞。
3.1.1 一个小问题:R读入数据后会改变列名
这个写了个推送,这里只列下结论:
- R认为短横线
-是无效的,所以如果读入的列名中包含了-,它会默认将其替换为.,目的是确保能及时检验是否出现重复的列名- 如果原本数据中的列名包括
-或者其他R不识别的字符,自己还想让读到R的数据与原数据保持一致,就需要使用check.names = FALSE不让read.csv或者read.delim读入的数据框中列名的-变成.
- plate1 <- read.delim(file.path(tempdir(), "counts_Calero_20160113.tsv"),
- header=TRUE, row.names=1, check.names=FALSE)
- plate1[1:2,1:2]
- # Length SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
- # ENSMUSG00000102693 1070 0
- # ENSMUSG00000064842 110 0
- dim(plate1)
- # [1] 46703 97
- plate2 <- read.delim(file.path(tempdir(), "counts_Calero_20160325.tsv"),
- header=TRUE, row.names=1, check.names=FALSE)
其中第一列是基因长度,把这个信息提取出来,就可以去掉啦
- gene.lengths <- plate1$Length # 提取基因长度
- plate1 <- as.matrix(plate1[,-1])
- plate2 <- as.matrix(plate2[,-1])
然后把两个细胞板的表达矩阵按列整合起来,但首先要检测它们的行名是否一致:
- stopifnot(identical(rownames(plate1), rownames(plate2)))
- all.counts <- cbind(plate1, plate2)
3.2 利用表达矩阵创建SCE对象
为了后续操作的流畅,这里将表达矩阵存储到一个SingleCellExperiment s对象中(scater、scran包使用的也是这个对象),这样可以添加许多行或列的注释信息
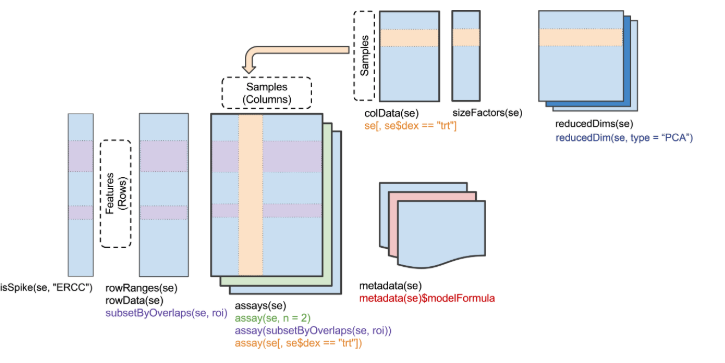
- suppressMessages(library(SingleCellExperiment))
- sce <- SingleCellExperiment(list(counts=all.counts))
- # 将基因长度信息添加在rowData中作为注释
- rowData(sce)$GeneLength <- gene.lengths
- sce
- # class: SingleCellExperiment
- # dim: 46703 192
- # metadata(0):
- # assays(1): counts
- # rownames(46703): ENSMUSG00000102693 ENSMUSG00000064842 ... SIRV7
- # CBFB-MYH11-mcherry
- # rowData names(1): GeneLength
- # colnames(192): SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
- # SLX-9555.N701_S503.C89V9ANXX.s_1.r_1 ...
- # SLX-11312.N712_S508.H5H5YBBXX.s_8.r_1
- # SLX-11312.N712_S517.H5H5YBBXX.s_8.r_1
- # colData names(0):
- # reducedDimNames(0):
- # spikeNames(0):
记得这个数据是包含ERCC的,所以要把这些外源的转录本提取出来,发现有92个ERCC,符合认知:
关于ERCC spike-in的小知识:
spike-in是已知浓度的外源RNA分子。在单细胞裂解液中加入spike-in后,再进行反转录。最广泛使用的spike-in是由External RNA Control Consortium (ERCC)提供的。目前使用的赛默飞公司提供的ERCC是包括92个不同长度和GC含量的细菌RNA序列,因此它和哺乳动物转录组不同,主要体现在转录本长度、核苷酸成分、polyA长度、没有内含子、没有二级结构。polyA尾大约15nt(一般保守的内源mRNA的polyA尾有250nt)。用它是为了更好地估计和消除单细胞测序文库的系统误差(除此以外,还有一种UMI在10X中常用)。ERCC应该在样本解离后、建库前完成添加。
- # grepl返回逻辑值
- isSpike(sce, "ERCC") <- grepl("^ERCC", rownames(sce))
- summary(isSpike(sce, "ERCC"))
- ## Mode FALSE TRUE
- ## logical 46611 92
除了ERCC,这个数据集还有点奇怪,因为它还包含了其他的spike-in信息,也就是Spike-In RNA Variants (SIRV)。为了操作简便,我们这里只用ERCC就好,这7个SIRV就可以舍去
- is.sirv <- grepl("^SIRV", rownames(sce))
- summary(is.sirv)
- ## Mode FALSE TRUE
- ## logical 46696 7
- sce <- sce[!is.sirv,]
来自作者的建议:
挑选ERCC,我们这里用的正则表达匹配:^ERCC ,但是要注意,人类基因组中也会有以ERCC开头的基因集,比如来自GeneCard的结果:
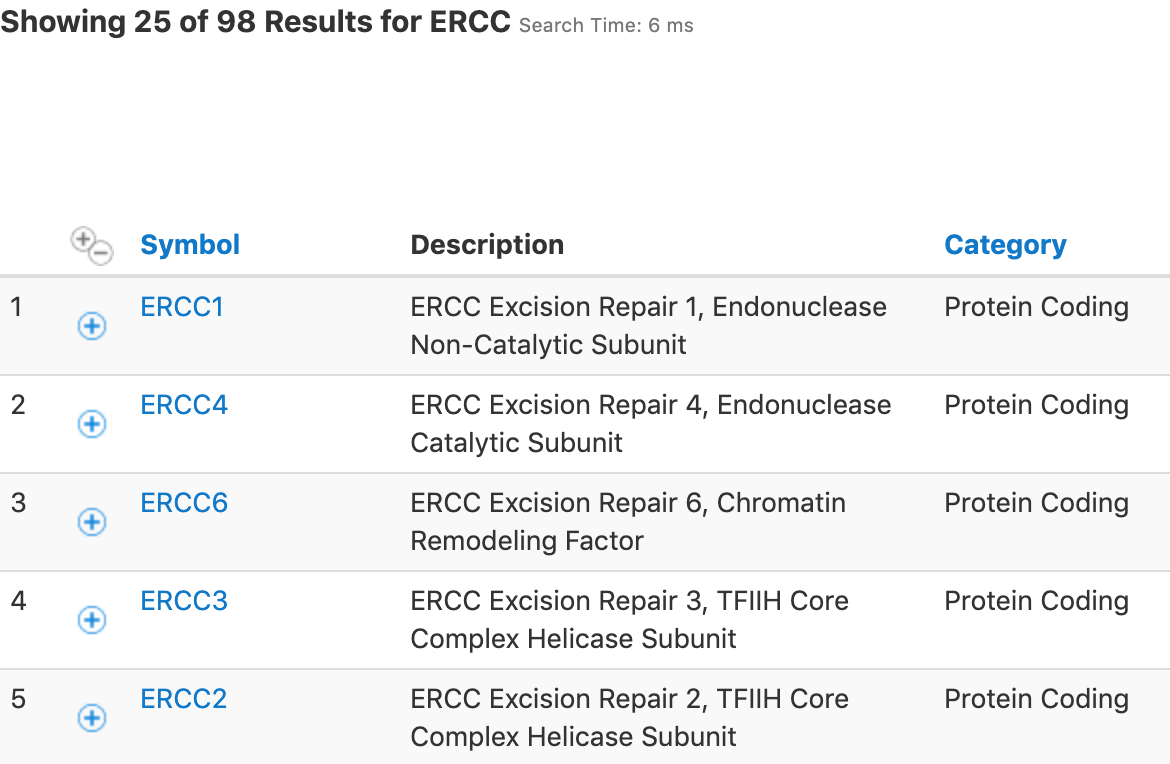
这样在挑选spike-in的时候,就可能会出错,因此在发布表达矩阵的时候,最好使用标准命名(Entrez、Ensembl),最好不要直接用Symbol命令
3.3 添加整合细胞注释信息
上面👆的操作是对基因的信息进行操作,接下来就是对细胞信息整合
3.3.1 首先保证注释信息与表达矩阵的细胞顺序一一对应
细胞的注释信息存储在sdrf.txt文件中,使用这个文件时需要注意:检查这个文件的行顺序是否与上面表达矩阵的列顺序一一对应(下面代码会有一个操作,保证二者顺序一致)。如果不一致,表达矩阵的细胞就会对应到错误的细胞注释信息。
- # 依旧使用check.names读入细胞注释信息
- metadata <- read.delim(lun.sdrf, check.names=FALSE, header=TRUE)
- metadata[1:3,1:3]
- # match的作用是返回前一个参数在后一个参数中的位置
- m <- match(colnames(sce), metadata[["Source Name"]])
- # 这样m就存储了一系列的位置,将表达矩阵的列和细胞注释信息的行顺序一一对应起来
- stopifnot(all(!is.na(m))) # 检查是否完整(即包含所有的细胞)
- metadata <- metadata[m,]
- # 这样就保证了我们只取到和表达矩阵相关的细胞注释,其他无用信息可以去除
- head(colnames(metadata))
- ## [1] "Source Name" "Comment[ENA_SAMPLE]"
- ## [3] "Comment[BioSD_SAMPLE]" "Characteristics[organism]"
- ## [5] "Characteristics[cell line]" "Characteristics[cell type]"
match函数:想按谁进行匹配,谁就放在第一个位置,比如最后想按表达矩阵细胞名进行匹配,让细胞注释细胞名”屈服于“表达矩阵细胞名的顺序,就要把表达矩阵细胞名放第一个
例如:B[match(A,B)]就实现了B按照A排列
3.3.2 然后添加细胞注释信息到对象
例如这里可以将细胞来源(也就是对应细胞板信息)、细胞表型添加一下,要注意它们都是因子型变量:
- # 先添加细胞来源
- colData(sce)$Plate <- factor(metadata[["Factor Value[block]"]])
- # 然后添加细胞表型
- pheno <- metadata[["Factor Value[phenotype]"]]
- levels(pheno) <- c("induced", "control")
- colData(sce)$Oncogene <- pheno
- # 最后看看新增的细胞表型数据
- table(colData(sce)$Oncogene, colData(sce)$Plate)
- ##
- ## 20160113 20160325
- ## induced 48 48
- ## control 48 48
3.4 添加整合基因注释信息
我们上游定量得到的基因一般都是Ensembl或Entrez的ID,这样保证了ID号的准确性和稳定性。但是,只看这样的ID我们还是不能知道这个是什么基因,因为常用的还是Symbol ID,像“TP53“、”BRCA1“等,因此就涉及到了另一个常用知识点:
3.4.1 基因ID转换
这里是小鼠数据,因此使用小鼠的注释包
第一种方法:mapIds(得到的是一个字符串),并且得到的基因名和原来矩阵的基因名是一一对应的
- library(org.Mm.eg.db)
- symb <- mapIds(org.Mm.eg.db, keys=rownames(sce), keytype="ENSEMBL", column="SYMBOL")
- # 看看数据类型
- > head(symb)
- ENSMUSG00000102693 ENSMUSG00000064842 ENSMUSG00000051951 ENSMUSG00000102851
- NA NA "Xkr4" NA
- ENSMUSG00000103377 ENSMUSG00000104017
- NA NA
- # 检测一下它们的Ensembl ID是否一样
- > identical(names(symb),rownames(sce))
- [1] TRUE
- rowData(sce)$SYMBOL <- symb
- rowData(sce)$ENSEMBL <- rownames(sce)
第二种方法:select(得到的是一个数据框),它得到的基因名顺序和原矩阵基因名顺序不同,需要再match
- library(org.Mm.eg.db)
- tmp <- select(org.Mm.eg.db, keys=rownames(sce), keytype="ENSEMBL", column="SYMBOL")
- # 看看数据类型
- > head(tmp)
- ENSEMBL SYMBOL
- 1 ENSMUSG00000102693 <NA>
- 2 ENSMUSG00000064842 <NA>
- 3 ENSMUSG00000051951 Xkr4
- 4 ENSMUSG00000102851 <NA>
- 5 ENSMUSG00000103377 <NA>
- 6 ENSMUSG00000104017 <NA>
- # 检测一下它们的Ensembl ID是否一样
- > identical(tmp$ENSEMBL,rownames(sce))
- [1] FALSE
- # 再match一下,按谁匹配谁放第一个
- > identical(tmp$ENSEMBL[match(rownames(sce),tmp$ENSEMBL)],rownames(sce))
- [1] TRUE
- # 和第一种方法的比价,结果就一样啦
- > identical(tmp$SYMBOL[match(rownames(sce),tmp$ENSEMBL)],as.character(symb))
- [1] TRUE
- # 于是第二种方法的结果就是
- rowData(sce)$SYMBOL <- tmp$SYMBOL[match(rownames(sce),tmp$ENSEMBL)]
- rowData(sce)$ENSEMBL <- rownames(sce)
综上两点,看来使用mapIds更加方便
好,最后看一下添加的基因信息:
- > head(rowData(sce))
- DataFrame with 6 rows and 3 columns
- GeneLength ENSEMBL SYMBOL
- <integer> <character> <character>
- ENSMUSG00000102693 1070 ENSMUSG00000102693 NA
- ENSMUSG00000064842 110 ENSMUSG00000064842 NA
- ENSMUSG00000051951 6094 ENSMUSG00000051951 Xkr4
- ENSMUSG00000102851 480 ENSMUSG00000102851 NA
- ENSMUSG00000103377 2819 ENSMUSG00000103377 NA
- ENSMUSG00000104017 2233 ENSMUSG00000104017 NA
3.4.2 整合行名(ID+Name)
上面👆的转Symbol name的操作确实很直观,但是毕竟不是所有的Ensembl ID都有Symbol name对应的。从上面的几行结果看到,没有Symbol 对应的就会被填成”NA“;另外如果出现重复的symbol,我们还要去重。这就用到scater包的一个功能:
uniquifyFeatureNames() make gene name unique and valid,如果有symbol name的,它会将Ensembl ID替换为symbol name,没有name的仍然使用ID;如果name相同、ID不同(即两个Ensembl ID对应同一个Symbol name),它会将ID与name组合保证特异性(详见
?uniquifyFeatureNames)
- library(scater)
- # 参数很简单,第一个是ID,第二是names
- rownames(sce) <- uniquifyFeatureNames(rowData(sce)$ENSEMBL, rowData(sce)$SYMBOL)
- head(rownames(sce))
- ## [1] "ENSMUSG00000102693" "ENSMUSG00000064842" "Xkr4"
- ## [4] "ENSMUSG00000102851" "ENSMUSG00000103377" "ENSMUSG00000104017"
3.4.3 添加基因所在染色体位置信息
利用这个包:TxDb.Mmusculus.UCSC.mm10.ensGene
可以参考:http://bioinfoblog.it/tag/txdb/
- BiocManager::install('TxDb.Mmusculus.UCSC.mm10.ensGene')
- library(TxDb.Mmusculus.UCSC.mm10.ensGene)
- # TxDb storing transcript annotations(包括CDS、Exon、Transcript的start、end、chr-location、strand)
- columns(TxDb.Mmusculus.UCSC.mm10.ensGene)
- # 返回一个GRanges对象
- head(transcripts(TxDb.Mmusculus.UCSC.mm10.ensGene,columns=c('CDSCHROM')))
- # 补充:如果是想看org.db其中的内容
- if(F){
- keytypes(org.Hs.eg.db)
- head(mappedkeys(org.Hs.egENSEMBL))
- }
- location <- mapIds(TxDb.Mmusculus.UCSC.mm10.ensGene,
- keys=rowData(sce)$ENSEMBL,
- column="CDSCHROM", keytype="GENEID")
- rowData(sce)$CHR <- location
- summary(location=="chrM")
- ## Mode FALSE TRUE NA's
- ## logical 22428 13 24255
结果会发现有大量的NA,然后比对到线粒体的有13个
4 重要的细胞质控
4.1 首先得到一系列细胞质控指标
过滤低质量的细胞目的是保证计数的误差不会影响下游的分析,一般重点看这几个QC结果:
- 文库大小(library size):每个样本/文库中全部feature(包括内源基因、spike-in等)的表达量。如果一个细胞文库太小,说明RNA信息没有被有效地捕获出来(也就是在文库制备时没有有效地反转录、扩增)
- 每个细胞中表达的feature数量:一个细胞中非0表达量的feature信息(内源基因、spike-in等)。如果一个细胞中有表达量的基因很少,整个基因覆盖度不高,还是说明很多转录本没有被有效捕获出来
- 比对到spike-in转录本的reads比例:分母是每个细胞文库的大小,分子就是细胞其中spike-in的数量。如果这个值很高,间接说明内源基因占比很低,细胞质量不好。
- 如果不使用spike-in的信息时,可以用线粒体基因来替代。高比例的线粒体基因同样也代表了细胞质量低(Islam et al. 2014; Ilicic et al. 2016)。原因如下:pre-mRNA在细胞核内完成一系列复杂加工后,成熟的mRNA被转运到细胞质中进行翻译。如果一个细胞破裂,大量的细胞质mRNA会外流,但是线粒体的体积比mRNA分子要大得多,因此最后剩下的线粒体mRNA也更多,最后经过反转录、扩增、比对到线粒体基因组的reads数也更多。【关于低质量细胞的影响,下面👇(4.3部分)还有介绍哦!】
对每个细胞利用scater包(McCarthy et al. 2017)的calculateQCMetrics()计算质控指标,结果会在行和列中增加metadata。
- # 之前ERCC已经使用isSpike()添加到sce中,这里再将另一个影响因素Mt添加进来
- mito <- which(rowData(sce)$CHR=="chrM")
- sce <- calculateQCMetrics(sce, feature_controls=list(Mt=mito))
- head(colnames(colData(sce)), 10)
- ## [1] "Plate" "Oncogene"
- ## [3] "is_cell_control" "total_features_by_counts"
- ## [5] "log10_total_features_by_counts" "total_counts"
- ## [7] "log10_total_counts" "pct_counts_in_top_50_features"
- ## [9] "pct_counts_in_top_100_features" "pct_counts_in_top_200_features"
4.1.1 然后做个可视化
根据可视化结果来推断哪些细胞是低质量的,比如文库小、feature表达量低、高spike-in/Mt占比...
这样的细胞可能会影响下游分析,例如这群低质量细胞可能会独自聚成一类,然后影响我们对细胞亚群判断。因此可以先用scater的multiplot()作图
- # 首先得到细胞类型和批次的组合信息
- sce$PlateOnco <- paste0(sce$Oncogene, ".", sce$Plate)
- multiplot(
- plotColData(sce, y="total_counts", x="PlateOnco"),
- plotColData(sce, y="total_features_by_counts", x="PlateOnco"),
- plotColData(sce, y="pct_counts_ERCC", x="PlateOnco"),
- plotColData(sce, y="pct_counts_Mt", x="PlateOnco"),
- cols=2)
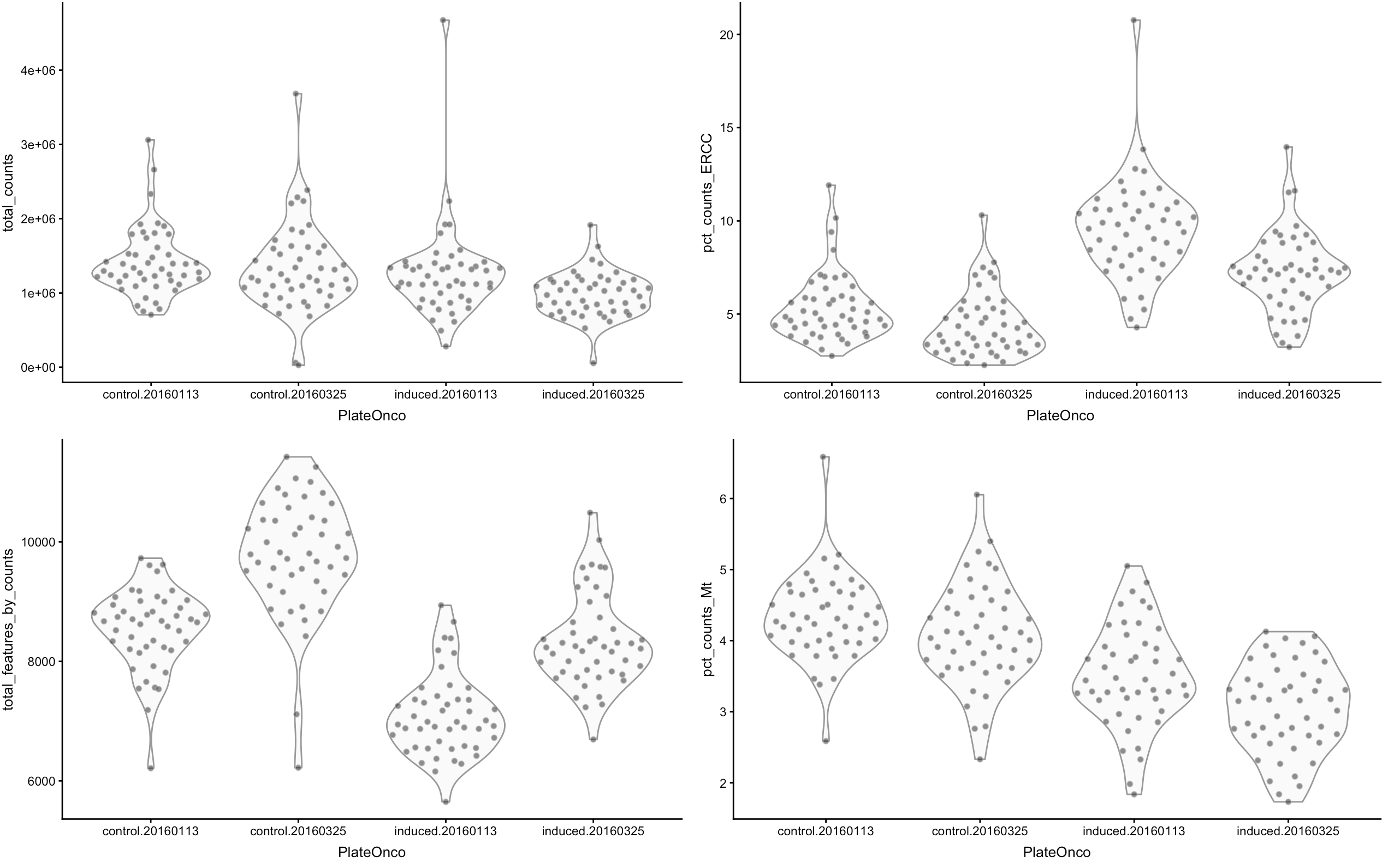
除了对每个统计指标分别可视化,还可以看两两指标之间的关系:
- par(mfrow=c(1,3))
- plot(sce$total_features_by_counts, sce$total_counts/1e6, xlab="Number of expressed genes",
- ylab="Library size (millions)")
- plot(sce$total_features_by_counts, sce$pct_counts_ERCC, xlab="Number of expressed genes",
- ylab="ERCC proportion (%)")
- plot(sce$total_features_by_counts, sce$pct_counts_Mt, xlab="Number of expressed genes",
- ylab="Mitochondrial proportion (%)")
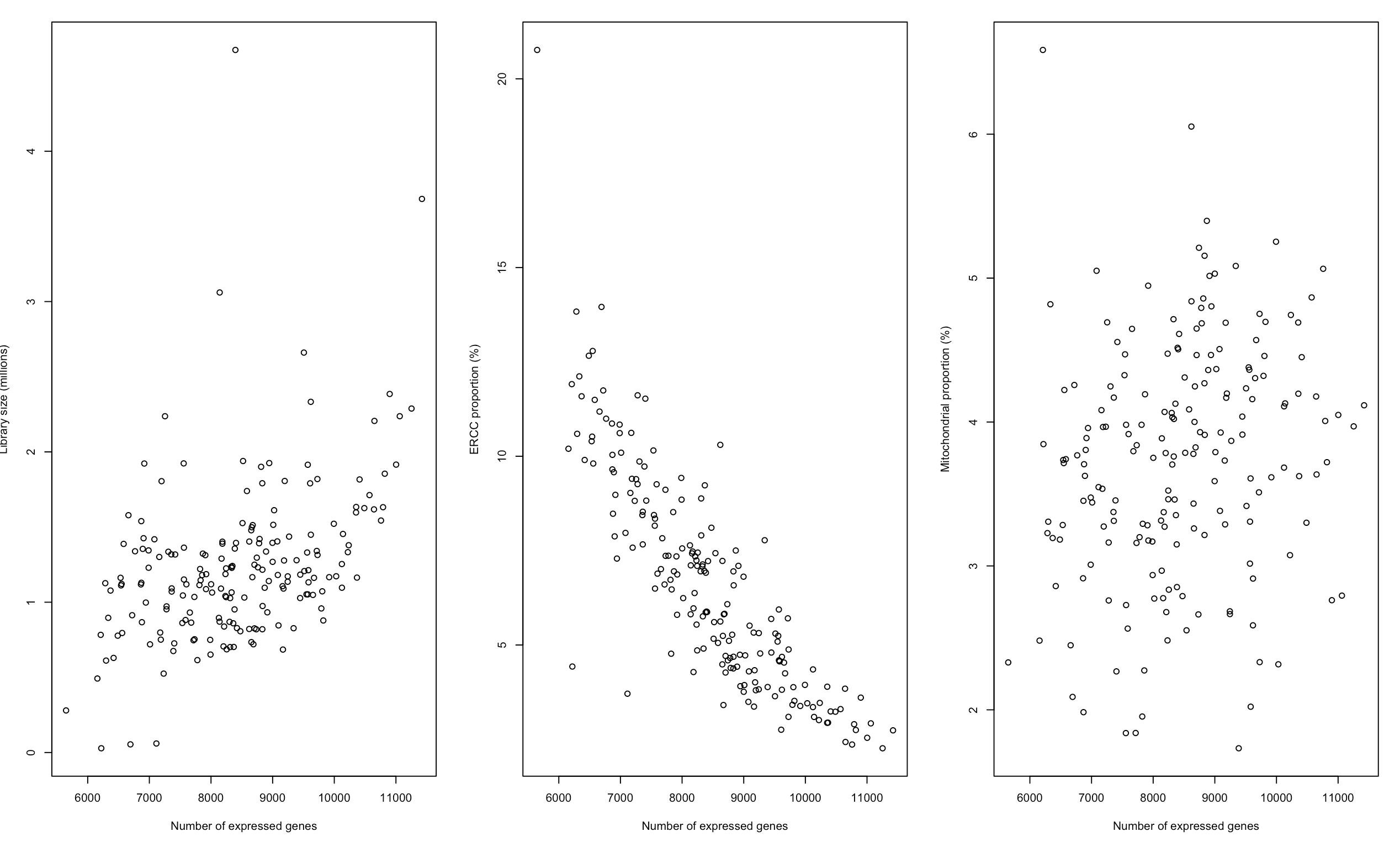
从上图就可以看到:
- 文库越大,表达基因一般也越多
- ERCC/MT越多,表达基因数量一般越少
4.2 为每个质控指标鉴定离群点
这个阈值的选择不要使用“一刀切”的模式,因为每种质控都有自己的范围,它们应该根据自己的特点去进行过滤。例如,不管细胞质量低不低,只要测序深,reads就多、有表达的feature数量也就多。还有,如果实验本身使用的spike-in RNA就多,那么最后得到的spike-in比例就高。我们过滤时就要先假设大部分细胞都是合格的,我们只需要剔除那些在QC结果中的离群点(Outliers)。
这些离群点的定义是根据绝对中位差(median absolute deviation,MAD),具体方法是:
Remove cells with log-library sizes that are more than 3 MADs below the median log-library size
看到使用了log归一化处理,它可以让非常大的值和非常小的值能够放在可以比较的范围之内。
下面过滤文库大小和有表达的feature数量(以低于中位数3倍MAD做为过滤条件)
- # 一般也就是去掉低文库大小、低表达基因、高spike-in
- libsize.drop <- isOutlier(sce$total_counts, nmads=3, type="lower",
- log=TRUE, batch=sce$PlateOnco)
- feature.drop <- isOutlier(sce$total_features_by_counts, nmads=3, type="lower",
- log=TRUE, batch=sce$PlateOnco)
- spike.drop <- isOutlier(sce$pct_counts_ERCC, nmads=3, type="higher",
- batch=sce$PlateOnco)
- 其中
batch参数是按照不同的批次(细胞类型+细胞板)分类统计离群点数量,毕竟看图4中不同批次组合得到的统计结果各不相同。另外,这里只用了spike-in作为过滤条件,这是因为线粒体本来在不同细胞中就有差别,而不一定是由于系统误差造成的,因此如果有spike-in就用它过滤就好。 - 另外,
isOutlier计算得到的阈值我们也能看到:- > attr(libsize.drop, "thresholds")
- control.20160113 control.20160325 induced.20160113
- lower 5.82883 5.622679 5.698215
- higher Inf Inf Inf
- induced.20160325
- lower 5.664173
- higher Inf
- > attr(spike.drop, "thresholds")
- control.20160113 control.20160325 induced.20160113
- lower -Inf -Inf -Inf
- higher 8.99581 8.105749 15.50477
- induced.20160325
- lower -Inf
- higher 12.71858
看下具体的过滤结果:
- keep <- !(libsize.drop | feature.drop | spike.drop)
- data.frame(ByLibSize=sum(libsize.drop), ByFeature=sum(feature.drop),
- BySpike=sum(spike.drop), Remaining=sum(keep))
- ## ByLibSize ByFeature BySpike Remaining
- ## 1 5 4 6 183
一共过滤了15个细胞,这里还比较正常(如果发现总体中有超过10%的细胞被过滤掉,那么就要考虑一下具体原因了。不过我认为这是对smart-seq2这种细胞数量较少的方法来讲的,10X的中五千细胞过滤后剩下两三千也是常见的)
接着,将原始的对象保存为416B_preQC.rds(下面的4.3部分会使用到这个原始对象~当然,这是可选部分)
然后进行过滤
- sce$PassQC <- keep
- saveRDS(sce, file="416B_preQC.rds")
- sce <- sce[,keep]
- dim(sce)
- ## [1] 46696 183
4.3【可选】对低质量细胞进行质控的必要性
来自:https://bioconductor.org/packages/3.9/workflows/vignettes/simpleSingleCell/inst/doc/qc.html#assumptions-of-outlier-identification
4.3.1 低质量细胞的影响
- 细胞破坏后,可能会导致线粒体或核RNAs占比升高(就是上面解释的大量细胞质中mRNA流失,而线粒体或核RNAs含量基本不变),很有可能会根据这个结果形成自己的一个个cluster
- 低质量的细胞一般文库比较小,而差异分析之前一般对文库大小进行一个归一化。比如正常细胞文库大小是100,某个基因表达量是2;损伤细胞的文库大小是10,这个基因表达量还是2。归一化后,损伤细胞中的这个基因表达量计算结果明显会高于正常细胞,呈现一种“本来不优秀,但班里人少了,排名就上升”的状态
- 细胞损伤可能会伴随RNA的流失,因此许多基因可能会被认为“下调”,尤其体现在细胞质核糖体RNA(另外还包括一些细胞质转录本)
- 影响方差估计和PCA结果。真实情况下,可能一个基因在两个细胞中差异并不显著,但是由于其中一个细胞质量低,导致基因表达量在这两个细胞中差异明显;反映在PCA结果就是:前几个主成分会抓取细胞质量的差异,因为这种差异体现得更明显,而将真正的生物学因素放到了后面几个主成分中,因此得到的PCA结果其实也只是反映了细胞质量的差异,而非真正的生物学差异
4.3.2 需要注意的点
- 如果一个细胞群体异质性较高,那么很有可能一些高质量细胞本身表达的数量就是比其他细胞少,但事实上它不是技术误差造成的。因此不能通过一个固定的阈值进行过滤,而要“因地制宜”,根据每群细胞各自的特性(比如各自的中位值),然后结合一定的统计指标(例如3倍的MAD)
- 过滤的细胞会不会属于某一个具有生物意义的细胞类群,如果真的是,那么就会有相应的marker基因高表达。
4.3.3 初步看一下
先做一个散点图,重点看看有没有基因明显向舍弃细胞方向偏移:
- # 计算丢弃和保留的细胞平均表达量(这里的平均表达量不是我们直接用apply+mean得出的结果,它计算了size factor
- # calcAverage具体操作如下:The size-adjusted average count is defined by dividing each count by the size factor and taking the average across cells. All sizes factors are scaled so that the mean is 1 across all cells, to ensure that the averages are interpretable on the scale of the raw counts.
- library(SingleCellExperiment)
- sce.full.416b <- readRDS("416B_preQC.rds")
- library(scater)
- lost <- calcAverage(counts(sce.full.416b)[,!sce.full.416b$PassQC])
- kept <- calcAverage(counts(sce.full.416b)[,sce.full.416b$PassQC])
- # 在上面得到的平均值中,将每个数都与平均值中(除0以外)最小的数进行比较,取最大的那个值作为最终的平均值
- capped.lost <- pmax(lost, min(lost[lost>0]))
- capped.kept <- pmax(kept, min(kept[kept>0]))
- plot(capped.lost, capped.kept, xlab="Average count (discarded)",
- ylab="Average count (retained)", log="xy", pch=16)
- is.spike <- isSpike(sce.full.416b)
- points(capped.lost[is.spike], capped.kept[is.spike], col="red", pch=16)
- is.mito <- rowData(sce.full.416b)$is_feature_control_Mt
- points(capped.lost[is.mito], capped.kept[is.mito], col="dodgerblue", pch=16)
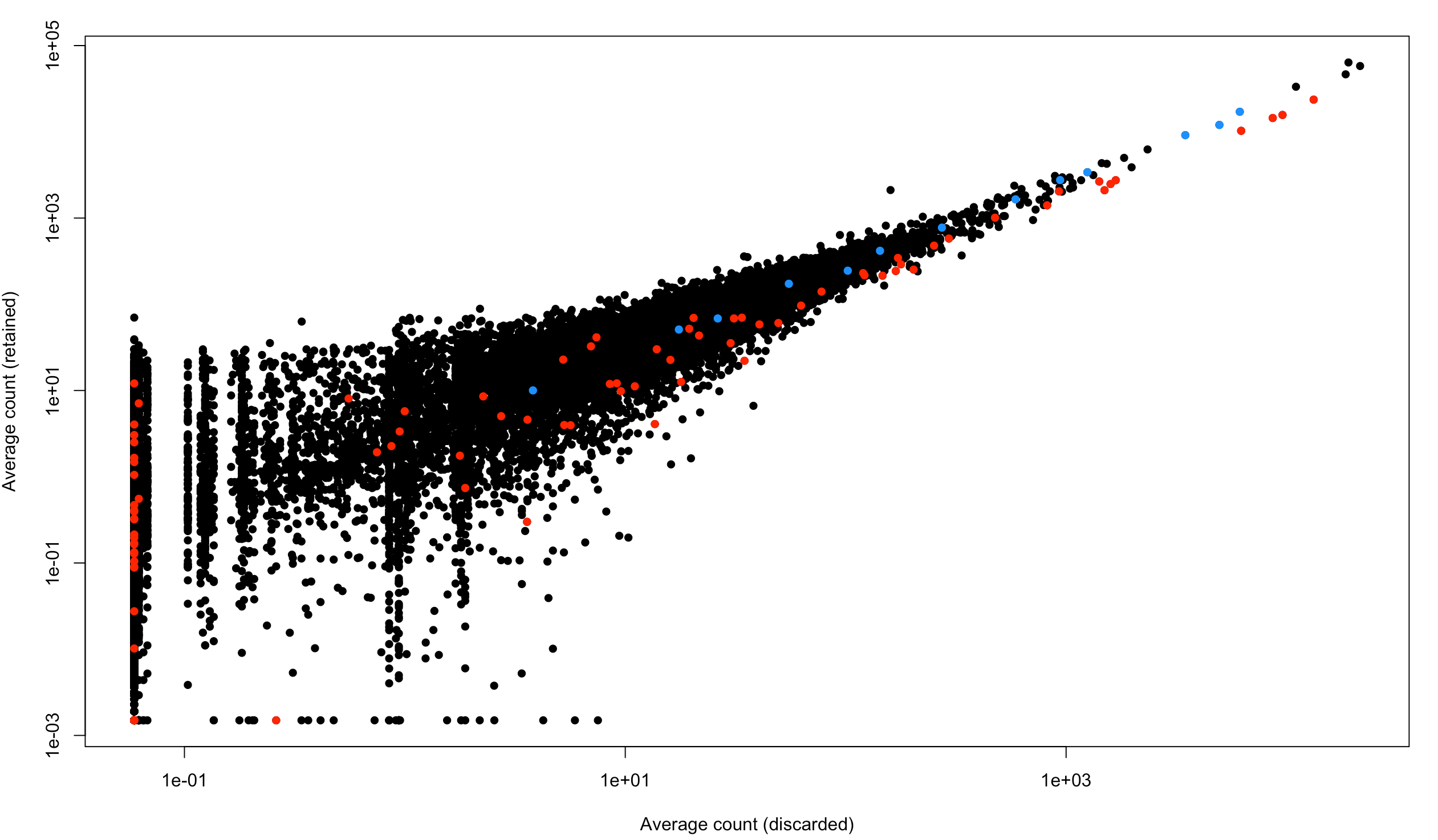
图中表示了舍弃的细胞和保留的细胞中基因平均表达量。每个点是一个基因,红色是spike-in,蓝色是线粒体基因
看到:整体方向还是偏向于y轴,虽然也有个别的点是偏向右下方的,下面👇就看看最偏右下方的那些点到底是不是marker基因?
4.3.4 认真再检查一下
方法就是:计算retain和discard两组细胞的基因表达量差异倍数( log-fold changes)。
核心就是:利用了edgeR的predFC函数,因为我们并不关心这两组之间的差异,这是想看看哪些基因在discard组更高表达一些。于是用这个函数分别根据每个组的表达量,再设置一个design实验设计矩阵就可以。没有正常做差异分析那么严格
- library(edgeR)
- #!! 重点在于DGEList中group的设置:数小的是control,也就是logFC的分母(自己也可以将group调换一下试试,结果看看是不是相反的)
- y <- cbind(lost, kept)
- y <- DGEList(y, group=c(2,1))
- design <- model.matrix(~group, data=y$samples)
- predlfc <- predFC(y, design)[,2]
- info <- data.frame(logFC=predlfc, Lost=lost, Kept=kept,
- row.names=rownames(sce.full.416b))
- head(info[order(info$logFC, decreasing=TRUE),], 20)
- # edgeR实验设计矩阵长这样:
- > design
- (Intercept) group2
- lost 1 1
- kept 1 0
- attr(,"assign")
- [1] 0 1
- attr(,"contrasts")
- attr(,"contrasts")$group
- [1] "contr.treatment"
结果就得到了:
- ## logFC Lost Kept
- ## ENSMUSG00000104647 6.844237 7.515034 0.000000000
- ## Nmur1 6.500909 5.909533 0.000000000
- ## Retn 6.250501 10.333172 0.196931018
- ## Fut9 6.096356 4.696897 0.010132032
- ## ENSMUSG00000102352 6.077614 9.393793 0.206637368
- ## ENSMUSG00000102379 6.029758 4.244690 0.000000000
- ## 1700101I11Rik 5.828821 4.483094 0.039199404
- ## Gm4952 5.698380 6.580862 0.172999108
- ## ENSMUSG00000106680 5.670156 3.389236 0.005234611
- ## ENSMUSG00000107955 5.554616 5.268508 0.132532601
- ## Gramd1c 5.446975 4.435342 0.103783669
- ## Jph3 5.361082 4.696897 0.139188080
- ## ENSMUSG00000092418 5.324462 3.395752 0.056931488
- ## 1700029I15Rik 5.316226 8.199510 0.394588776
- ## Pih1h3b 5.307439 2.546814 0.000000000
- ## ENSMUSG00000097176 5.275459 2.541927 0.003772842
- ## Olfr456 5.093383 2.186536 0.000000000
- ## ENSMUSG00000103731 5.017303 3.315016 0.107148909
- ## Klhdc8b 4.933215 19.861036 1.635081878
- ## ENSMUSG00000082449 4.881422 1.878759 0.000000000
挑出来top20在discard中相对高表达的基因,但其中并没有marker基因,这就足够了。不用关心logFC的实际数值,眼前的高可能是因为细胞破损后,计算得到的线粒体基因、细胞质核糖体基因或者核RNA占比升高,计算出来的的表达量也升高,因而比正常的细胞要高,不过这是一种“虚假繁荣”的假象而已。
4.3.5 如何避免丢失一些细胞类型
最直接的方法就是修改(上面4.2中isOutlier函数的nmads=参数) ,另外如果我们知道哪些细胞属于什么细胞类型,也可以提前定义出来避免过滤掉。最粗暴的还是不进行任何的细胞过滤,这样虽然确保了不会丢失,但同时增加了无用细胞的占比。因此,要不要过滤、怎么过滤还是要取决于个人的安排。
不得不说,得到的细胞质量和细胞类型还真有一定的关系。如果某种细胞就是在细胞提取阶段不配合,导致后来检测到这群细胞都有损伤,最好还是在保证实验设计的前提下去掉它们=》“牺牲小我保存大我”
4.3.6 其他一些质控方法
- 使用固定的阈值:比如设置文库大小阈值为100000,表达基因数量4000。但是这些都需要具有实际的经验,最好还是不要乱设置这些值,因为很有可能会由于自己的随意发挥,而抛弃了一些珍贵的细胞类型
- 看基于质控结果进行PCA分析的离群点: 前期会对每个细胞统计总reads数、总基因数等6个指标,然后用这些指标进行PCA。具体
runPCA的设置如下图:
如果PCA的结果有离群点,就说明在这些指标中(不论太大或太小)某些细胞是“脱离群众的”。举个例子:
- sce.tmp <- runPCA(sce.full.416b, use_coldata=TRUE, detect_outliers=TRUE)
- ##
- ## FALSE TRUE
- ## 187 5
- 另外还可以参考2019年出的cellity包
4.3.7 一个“太随意”的错误案例
例如:在没有任何先验知识情况下,我们直接对一个单细胞对象中一个统计指标(total_counts)设置一个阈值(1000)进行过滤:
- wrong.keep <- sce.pbmc$total_counts >= 1000
- lost <- calcAverage(counts(sce.pbmc)[,!wrong.keep])
- kept <- calcAverage(counts(sce.pbmc)[,wrong.keep])
- # 然后同上面一样做一个散点图
- # Avoid loss of points where either average is zero.
- capped.lost <- pmax(lost, min(lost[lost>0]))
- capped.kept <- pmax(kept, min(kept[kept>0]))
- plot(capped.lost, capped.kept, xlab="Average count (discarded)",
- ylab="Average count (retained)", log="xy", pch=16)
- platelet <- c("PF4", "PPBP", "SDPR")
- points(capped.lost[platelet], capped.kept[platelet], col="orange", pch=16)
很明显,这个图有点向下偏,也就是说有一些基因在discard组中表达量更多。然后测试了3个血小板相关的基因(橘黄色),发现它们确实在discard组中表达量更高,说明其中有部分血小板细胞被丢掉了
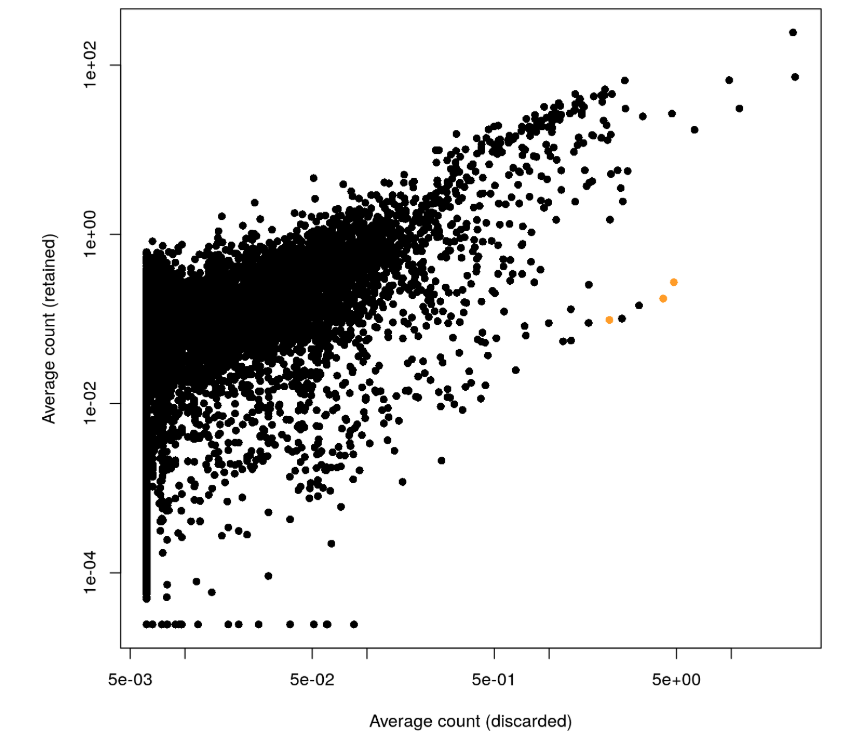
接着用logFC再检验一下:
- coefs <- predFC(cbind(lost, kept), design=cbind(1, c(1, 0)))[,2]
- info <- data.frame(logFC=coefs, Lost=lost, Kept=kept,
- row.names=rownames(sce.pbmc))
- head(info[order(info$logFC, decreasing=TRUE),], 20)
- ## logFC Lost Kept
- ## PF4 6.615671 4.2289086 0.17421441
- ## PPBP 6.495522 4.8409530 0.27144834
- ## HIST1H2AC 6.291680 3.1195253 0.14435051
- ## GNG11 6.162314 2.5132359 0.10103056
- ## SDPR 5.950336 2.1430640 0.09754273
- ## TUBB1 5.610679 1.6478079 0.08989034
- ## CLU 5.462238 1.3202347 0.05559122
- ## ACRBP 5.325560 1.1933566 0.05436568
- ## NRGN 5.118699 1.3155255 0.12992550
- ## RGS18 5.009974 1.6512487 0.25377562
- ## MAP3K7CL 4.898185 0.9950268 0.08957073
- ## SPARC 4.646432 0.6559761 0.02487467
- ## MMD 4.616159 0.7450948 0.06367575
- ## PGRMC1 4.503524 0.7335820 0.08248177
- ## CMTM5 4.166574 0.4469937 0.01643304
- ## TSC22D1 4.120268 0.5128047 0.05920195
- ## HRAT92 4.116163 0.4214188 0.01144488
- ## GP9 4.091146 0.4610813 0.03703803
- ## CTSA 3.986470 0.8294337 0.27097756
- ## MARCH2 3.966528 0.5655020 0.12228759
发现,血小板几个相关基因的确在discard组中更高,说明之前按照sce.pbmc$total_counts >= 1000的过滤是错误的。而不检查有没有先验知识的话,后面分群我们也得不到这部分细胞
5 细胞周期分类
这里根据Scialdone et al. (2015) 提供的预测方法,简而言之就是利用一个做好的训练数据集和已知表达矩阵基因表达量变化进行分类。在训练数据集中,已经计算好了两两基因的差异(基因对,pair of genes),并且将属于不同细胞周期(它规定了3种量化水平:G1、S、G2M)且存在差异的基因对作为一个marker pair。然后就在已知表达矩阵中对每个细胞测试这些marker pairs与训练数据集中的相似程度,每个细胞最后都得到了在G1、S、G2/M水平的分值,最后根据分值将细胞归类。
主要利用了scran包中的cyclone函数
cyclone函数主要需要三个元素:一个是sce单细胞对象,一个是pairs参数,还有就是gene.names参数。第一个已准备好,第二个参数的意思可以看帮助文档,第三个参数要求是Ensembl ID

- # scran包安装好后,会在exdata文件夹中找到附件文件
- library(org.Mm.eg.db)
- # syste,.file会列出文件所在的路径,下图就是exdata文件夹下的文件,看到除了小鼠还有人的相关的RDS数据。这个RDS其实和平常看到的Rdata差不多,只不过Rdata是针对多个对象,Rds是针对一个对象进行存储和读取
- mm.pairs <- readRDS(system.file("exdata", "mouse_cycle_markers.rds",
- package="scran"))
- # 举个例子
- > head(mm.pairs$G1)
- first second
- 1 ENSMUSG00000000001 ENSMUSG00000001785
- 2 ENSMUSG00000000001 ENSMUSG00000005470
- 3 ENSMUSG00000000001 ENSMUSG00000012443
- 4 ENSMUSG00000000001 ENSMUSG00000015120
- 5 ENSMUSG00000000001 ENSMUSG00000022033
- 6 ENSMUSG00000000001 ENSMUSG00000023015
注意:这里小鼠的训练数据集是利用胚胎干细胞数据得到的,但对于其他细胞类型也是准确的 (Scialdone et al. 2015),可能是由于细胞周期相关转录的保守性 (Bertoli, Skotheim, and Bruin 2013; Conboy et al. 2007)。
具体的使用就很简单:
- system.time(assignments <- cyclone(sce, mm.pairs,
- gene.names=rowData(sce)$ENSEMBL))
- ## user system elapsed
- ## 21.740 0.376 26.856
- save(sce,assignments,file = '416B_cell_cycle.Rdata')
这个结果并不是说某个细胞一定就处于哪个细胞周期,它也是根据大样本量背景进行概率估计,然后计算一个分值,分值越高,说明某个细胞更有可能属于哪个细胞周期,给我们一定的参考。
例如,看一下G1和G2/M的分值情况,其中每个点代表一个细胞:
- plot(assignments$score$G1, assignments$score$G2M,
- xlab="G1 score", ylab="G2/M score", pch=16)
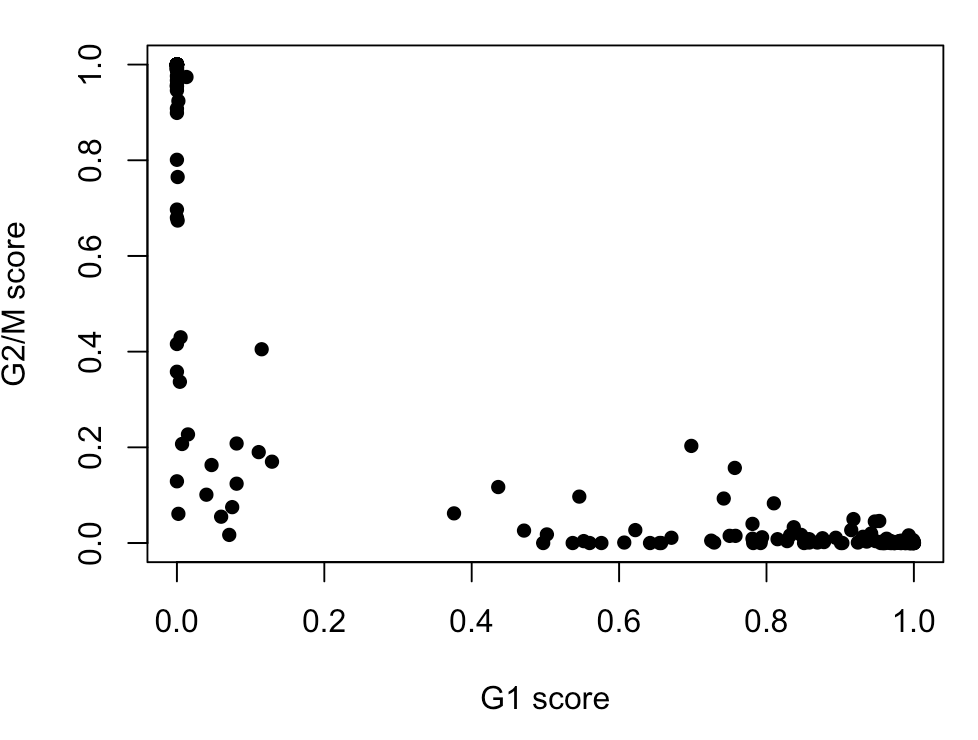
计算完细胞分值,就该划分细胞类型了
具体规则是:如果一个细胞在G1中得分大于0.5,并且它高于G2/M的得分,那么这个细胞就被划分到G1期;如果细胞在G2/M中得分大于0.5,并且高于G1的得分,那么它就划为G2/M期;如果细胞的G1、G2/M得分都不大于0.5,那么它就划为S期。于是上面👆的结果就可以被划分成:
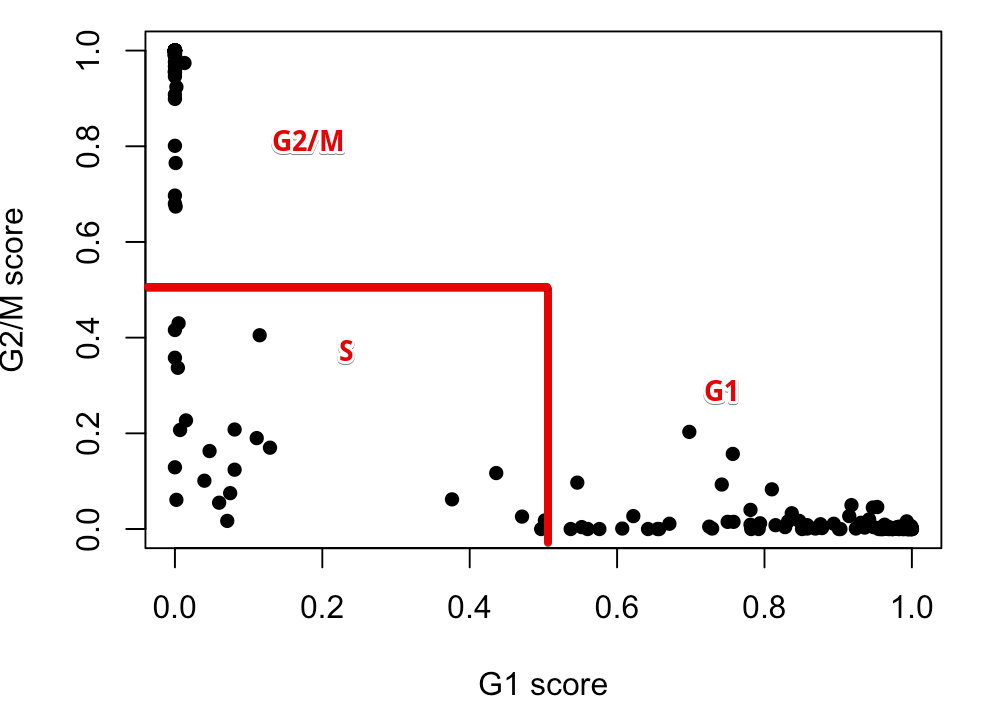
这里只是说明一下原理, 其实函数已经为我们划分好,存到了结果中:
- sce$phases <- assignments$phases
- table(sce$phases)
- ##
- ## G1 G2M S
- ## 98 62 23
作者的建议
- 为了去掉细胞周期带来的影响,我们一般只要某一群特定周期的细胞(一般是G1期)进行下游分析。当然如果其他时期的细胞数量不会造成明显的影响,我们也可以带着它们,到下游分析时直接将
assignments$phases作为一个批次效应因素考虑即可,这样既避免了其他周期细胞的干扰,又能避免丢失部分信息。 - 训练数据集虽然说对多种细胞类型都支持,如果本身的数据与训练数据相差太大(比如使用了不同的方法得到的数据),那么我们可以根据自己的数据去DIY一个分类器。使用
sandbag函数即可,同样如果研究其他的物种没有给定的分类器,我们就可以这样操作 - 在使用
cyclone之前不要过滤低丰度转录本。即使一个基因在任何一个细胞都不表达,但在周期推断环节这个基因名还是有作用的。因为这一步做的是一个基因对比较,所以它依然可以提供信息。
6 基因层面检查
6.1 检查高表达基因
目的是看看实验设计如何,是否在文库制备、比对环节出了问题
先看下高表达基因(例如top50)的ID。通常来讲,其中应该存在许多连续表达的转录本,比如核糖体或线粒体相关。如果出现了与实验预期不相符的基因,就应该关注一下。例如存在许多的ERCC转录本,就说明在文库制备中添加了太多的spike-in RNA,如果核糖体基因缺失或出现了一些假基因,那么可能在比对过程中选择了不太好的比对结果。
- # 这个代码可以借鉴,调整字体大小的参数
- fontsize <- theme(axis.text=element_text(size=12), axis.title=element_text(size=16))
- plotHighestExprs(sce, n=50) + fontsize

6.2 过滤低丰度基因
定义
低丰度基因就是表达量为0或接近0,在下游分析中基本起不到什么统计作用的基因。在假设检验中,它们不能提供足够的证据去推翻原假设,但同时它们的存在会增加多重检验校正的计算量。另外还有一个弊端,可能用英文的解释比较好理解:The discreteness of the counts may also interfere with statistical procedures, e.g., by compromising the accuracy of continuous approximations。因此下游分析之前,一般是要先去除低丰度基因。
作者认为“最佳的过滤方法”是:并非简单采用一次过滤应用于整个分析,而是每一步根据需要去过滤。他的理由是
A more aggressive filter is usually required to remove discreteness (e.g., for normalization) compared to that required for removing underpowered tests. For hypothesis testing, the filter statistic should also be independent of the test statistic under the null hypothesis (Bourgon, Gentleman, and Huber 2010)
利用平均表达量进行过滤
有许多指标可以定义低丰度,其中最常用的就是平均表达量,利用calcAverage函数,同时它还会根据各个文库大小进行一个校正(默认设置use_size_factors=TRUE)
- # 这里就只是简单统计一个平均值
- ave.counts <- calcAverage(sce, use_size_factors=FALSE)
- # 一个很有用的操作,就是如何在坐标轴上显示下标,看xlab如何设置的就知道了
- hist(log10(ave.counts), breaks=100, main="", col="grey80",
- xlab=expression(Log[10]~"average count"))
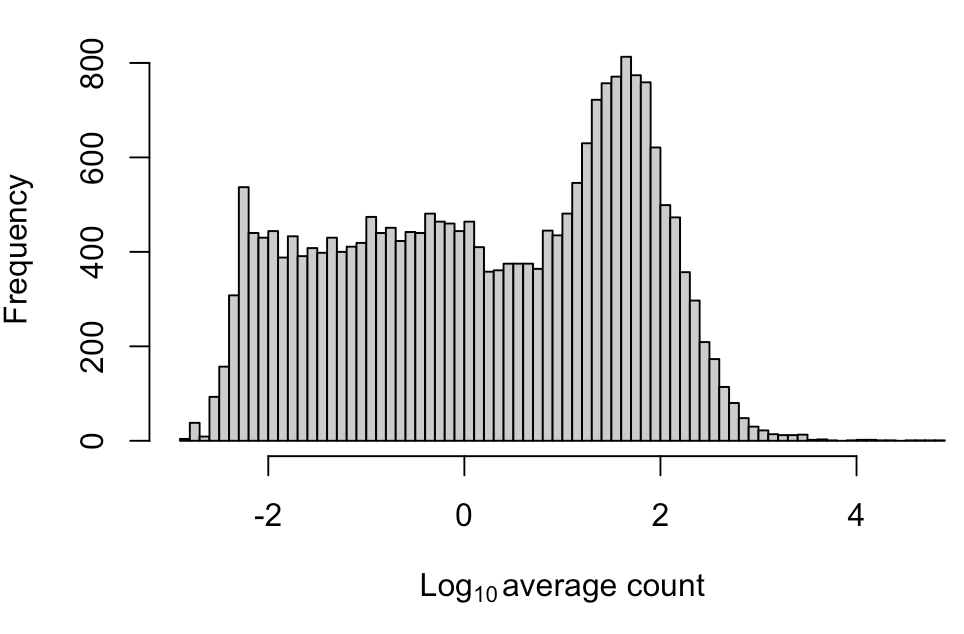
图中会观察到一个“山峰”在一片“高原”上拔地而起,这个“高原”就是大量的低丰度基因,我们过滤的话一般去掉平均表达量在1以下的,也就是图中横坐标0左边的区域
- demo.keep <- ave.counts >= 1
- filtered.sce <- sce[demo.keep,]
- summary(demo.keep)
- ## Mode FALSE TRUE
- ## logical 33490 13206
- # 可以看到去掉了3w多基因,可以考虑换一种方式,想办法多留一些基因
利用每个基因在多少细胞中表达进行过滤
这个指标和上面平均表达量是紧密相关的,因为一个基因在许多细胞中都表达,也意味着它的平均表达量不会太低,就像下面这张图中表示的一样。
- num.cells <- nexprs(sce, byrow=TRUE)
- smoothScatter(log10(ave.counts), num.cells, ylab="Number of cells",
- xlab=expression(Log[10]~"average count"))
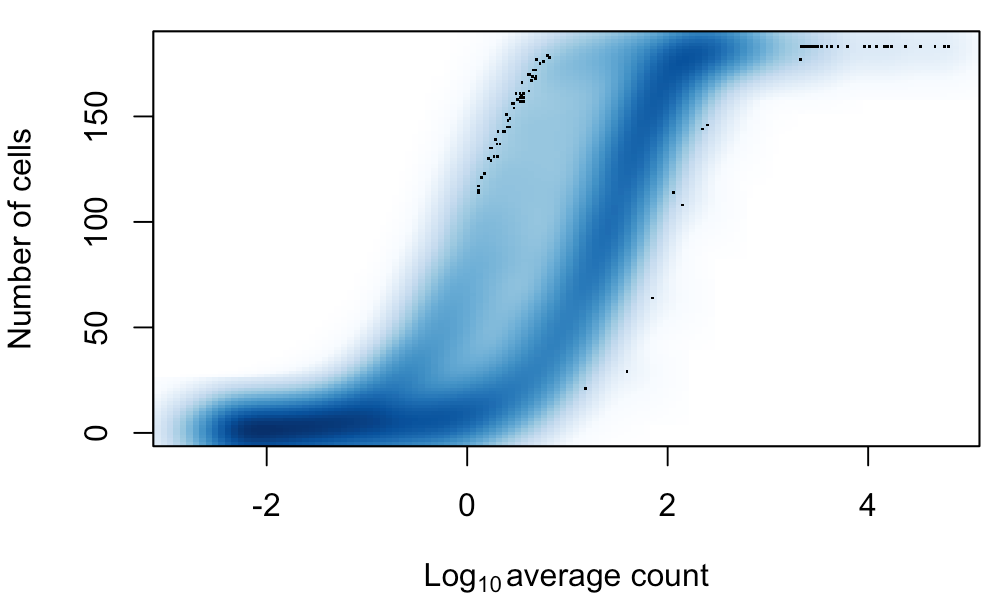
如果发现某个基因在很少细胞中表达,一般对这个基因是不感兴趣的,因为它很有可能是由于扩增偏差产生的“假表达量”。不过更严谨考虑,这个基因也可能就只是存在于某个罕见的细胞群体,而这个群体本身数量就很少。
这里选择宽松的过滤条件:一个基因至少在一个细胞中有表达,这个基因就留下
- to.keep <- num.cells > 0
- sce <- sce[to.keep,]
- summary(to.keep)
- ## Mode FALSE TRUE
- ## logical 22833 23863
- # 这样剩下2w多个基因
7 对细胞文库差异进行normalization
再次借助https://github.com/satijalab/seurat/issues/950的一个回答:
- Normalization "normalizes" within the cell for the difference in sequenicng depth / mRNA thruput
- Scaling "normalizes" across the sample for differences in range of variation of expression of genes
关于它们的中文翻译实在是错综复杂,记不清楚就简单记,反正没有正确答案,自己知道这一步做了什么最重要。normalization一般是对文库处理,目的消除一些技术差异;scale一般对基因表达量处理(典型的z-score:表达量减均值再除以标准差),目的是后续分析不受极值影响(比如做热图我们不关心哪个基因表达量最多,而关心哪个基因在样本间最有差异,虽然这个基因不一定表达量很高。比如A基因在样本1中表达量为900,在样本B中表达量为901;而B基因在样本1中表达量为10,在样本B中表达量为50。我们更关心B基因。但直接作图,往往会因为A基因的绝对表达量很高,而将B基因在图中掩盖掉。scale这个操作就是帮我们”拨开云雾见青天“)
7.1 去卷积方法(deconvolution)根据内源基因计算size factor
表达量的差异除了生物学因素外,还会受到细胞捕获效率、测序深度的影响(Stegle, Teichmann, and Marioni 2015)。在进行下游定量分析之前,需要利用归一化去掉这些“无关因素”产生的细胞差异。
假设大部分基因在细胞间并非差异表达,统计思想是:any systematic difference in count size across the non-DE majority of genes between two cells is assumed to represent bias and is removed by scaling.
更具体说就是计算“size factors” (represent the extent to which counts should be scaled in each library)
size factor的计算方法有多种,例如常规转录组中DESeq2的estimateSizeFactorsFromMatrix 、edgeR的calcNormFactors 。但是这里单细胞数据又和常规转录组不同,它存在了太多的低丰度甚至是0表达(从上面的过滤结果也能感知),一个细胞一个细胞单独进行处理应该是误差较大。
于是开发了一种基于去卷积的新方法:先将许多细胞的counts混合起来,来增加总体的counts值,提高了后续计算size factor的准确度 (Lun, Bach, and Marioni 2016)。计算得到的混合size factor(Pool-based size factors),再“去卷积”,将pool size factor还原给每个细胞的factor。
说起来复杂,操作简单:
- sce <- computeSumFactors(sce)
- summary(sizeFactors(sce))
- ## Min. 1st Qu. Median Mean 3rd Qu. Max.
- ## 0.3464 0.7117 0.9098 1.0000 1.1396 3.5695
如何知道计算的size factor准确性好不好呢?
做一个散点图,看看size factor与lib size的关系
- plot(sce$total_counts/1e6, sizeFactors(sce), log="xy",
- xlab="Library size (millions)", ylab="Size factor",
- col=c("red", "black")[sce$Oncogene], pch=16)
- # 右下角添加图例,对数据中两种细胞类型进行了区分
- legend("bottomright", col=c("red", "black"), pch=16, cex=1.2,
- legend=levels(sce$Oncogene))
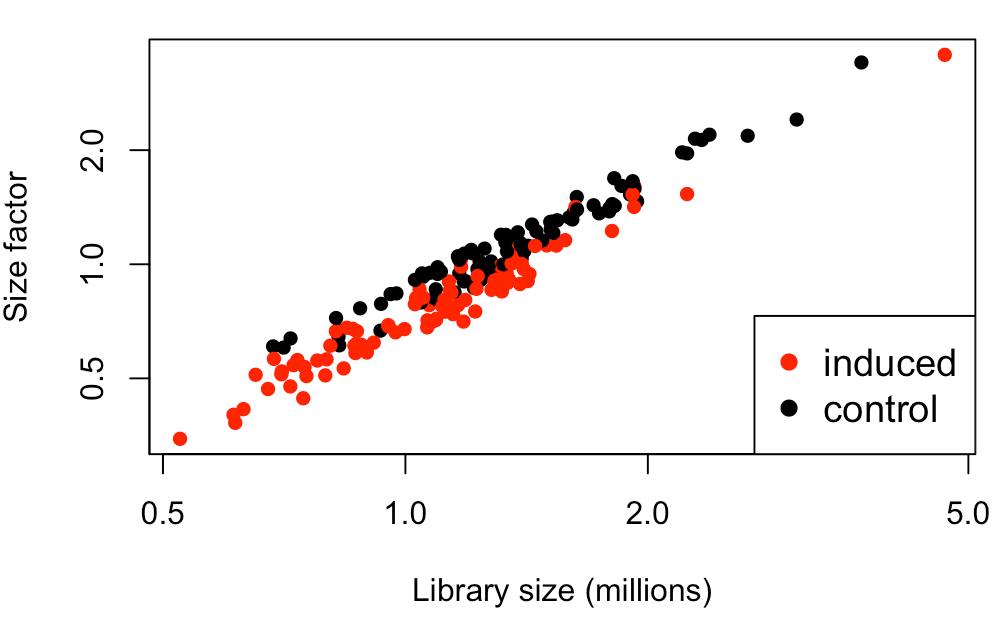
图中可以看到,size factor与文库大小相关性很好,说明大部分文库大小差异都是由于技术因素(捕获效率、测序深度)造成的。
【问:为何size factor不考虑生物因素?
答:因为这个名词创造之前,就已经做了无差异基因的假设条件,它只体现一些技术误差导致的差异,所以只有存在技术误差时,lib size才和size factor的相关趋势接近直线】
另外,从这个图中,看到红点(原癌基因诱导细胞)比黑点(对照组)的size factor要小,这从生物学角度也能解释通:黑点红点总体lib size一样,红点诱导后基因表达量升高(即生物学因素导致的文库变化升高),那么红点非生物学因素导致的文库变化就要降低
作者的观点
- 虽然去卷积法对高频次的0表达量处理效果不错,但如果太多的0表达量最后还是会失败。如果计算的size factor为负数,这很明显没有意义。因此
computeSumFactors函数在计算size factor前,会自动去先按平均表达量去除低丰度转录本,一般会以min.mean=1作为过滤条件 - 在标准化之前,细胞应该经常查看一下质控结果,保证去掉那些只有很少基因表达的细胞。否则,
computeSumFactors就会由于低质量细胞产生负数。因为size factor的计算涉及到了混合细胞的操作,最后操作的是pool library,因此每个细胞都有太多的0,最后校正也很难去掉它们 computeSumFactors()函数有一个参数sizes,它指定每个混池使用多少细胞计算size factor。每个混池使用的细胞数量越多,越准确,但也会消耗计算时间和内存。一般来讲,一个混池至少要用20个以上细胞才能保证有足够的非0表达量去计算。- 对于高度异质性的数据集来说,因为要保证”没有差异基因“这个原假设。所以可以先利用
quickCluster()做一个粗略的聚类,然后将结果通过clusters参数传递给computeSumFactors()再计算
7.2 根据spike-in转录本计算size factor
根据内源基因count结果计算的size factor是不适用于spike-in的,原因如下:
一般单细胞实验都不是定量实验(也就是说,我们事先不能规定好所有的细胞文库大小都是一样的),我们常常得到的数据是:包含更多转录本的细胞具有更大的文库,因此计算的size factor也更大,目的就是消除这个文库差异的影响。
这一点也很好理解,但是基于这个事实,我们人工在不同细胞中加入的spike-in转录本浓度却是一样的,在小文库细胞中某个ERCC表达量是100,换到另一个大文库细胞中,这个ERCC的表达量也只是近似,不会呈现数量级的差别。再用原来的方法对一个稳定的表达量进行校正,结果会”矫枉过正“。另外,即使使用了定量的实验,每个细胞的cDNA文库大小都是不变的,那么其中如果内源基因表达量升高,那么势必会导致外源spike-in表达量下降,它们的变化趋势不同,因此利用内源基因计算的size factor也不能直接利用到spike-in身上。
虽然原来方法不适用,但内源基因们都校正过了,spike-in也不能不校正。于是有一个单独为它开发的方法:假设所有的spike-in转录本表达无差异(当然这个假设是符合客观事实的)。 For each cell, the spike-in-specific size factor is defined as the total count across all transcripts in the spike-in set
利用一个函数:
- sce <- computeSpikeFactors(sce, type="ERCC", general.use=FALSE)
利用参数general.use=FALSE 将结果单独存放在sce对象中,保证了计算的size factor只对ERCC使用,而对内源基因生效
7.3 将size factor应用到normalization这一步
函数很简单:
- sce <- normalize(sce)
它做的事情就是:每个细胞中基因的表达量除以这个细胞计算的size factor,其中内源基因除以computeSumFactors()得到的factor,spike-in除以computeSpikeFactors()得到的factor。然后做个log2( +1)处理,降低数据的维度又不破坏数据原始结构(也就是说,原来大的数log完了还是大,但是在坐标轴上的数字就能减小,更方便展示数据全貌)
最后计算结果保存在:logcounts中,可以用logcounts(sce)调用表达矩阵
8 模拟基因表达中的技术噪音
我们在表达矩阵中观察到的基因表达量变化,一方面是由于真实的生物差异(也是我们关心的),另一方面可能就是技术噪音(我们并不关心)。为了区分这两种可能因素,需要从每个基因表达量差异提取出技术层面因素,构建一个模型方便观察。
方法就是:利用人工添加的定量的外源RNA(spike-in RNA),它是不存在第一种因素(真实生物差异)的,所以它们的变化可以直接反映技术噪音,然后将整体的内源基因平均表达量变化与spike-in进行拟合。
- 先利用
trendVar函数=》fit a mean-dependent trend to the variances of the log-expression values for the spike-in transcripts。它接受的数据是:normalized log-expression values, where each column corresponds to a cell and each row corresponds to a spike-in transcript另外设置一个参数block=来封闭某些已知的技术差异(比如我们已知的两个细胞板批次),进而探索更真实的技术噪音。这个函数根据基因的平均表达量 - 上面利用spike-in对所有基因得到了拟合值,它可以被认为是技术误差导致的方差,然后利用
decomposeVar()从总体方差中减去技术方差,得到的就是生物因素导致的方差
- var.fit <- trendVar(sce, parametric=TRUE, block=sce$Plate,
- loess.args=list(span=0.3))
- var.out <- decomposeVar(sce, var.fit)
- > head(var.out,2)
- DataFrame with 2 rows and 6 columns
- mean total
- <numeric> <numeric>
- ENSMUSG00000103377 0.00807160215879455 0.0119218654846046
- ENSMUSG00000103147 0.0346526071354525 0.0722196159042111
- bio tech
- <numeric> <numeric>
- ENSMUSG00000103377 -0.0238255786081426 0.0357474440927472
- ENSMUSG00000103147 -0.0812680860365811 0.153487701940792
- p.value FDR
- <numeric> <numeric>
- ENSMUSG00000103377 1 1
- ENSMUSG00000103147 0.999999999992144 1
作图检查一下,在下图👇中,每个点都是一个基因,画出了它们的表达量均值、技术因素方差、总体方差。
- 红点就是利用一个个spike-in RNA的表达量均值和方差画的
- 蓝色线条就是根据spike-in的表达量,利用
trendVar得到的拟合曲线,可以理解成技术因素的方差随着表达量的变化; - 黑色点是内源基因的总方差随表达量的变化,真正的生物因素导致的方差可以用黑点的纵坐标减去对应蓝线上的纵坐标,也就是蓝线上方的点都代表了真实的生物因素导致的差异。注意到蓝线以下还有点,难不成它们的生物因素差异为负? 还真的是,看上面的
head(var.out,2)结果,在bio这一列中真的存在负值,这个实际上是没有意义的,负值的产生一般是因为表达这个基因的细胞数量太少,结果估算不准确 - 除此以外,看到当表达量均值(横坐标)从0开始上升时,方差也在线性上升;当表达量高到一定程度,说明这种基因在细胞中分布就比较均衡(属于少数”表达量土豪级别并且实现了共同富裕“),因此取样估算过程的误差也降低【可以试想一下管家基因】
- plot(var.out$mean, var.out$total, pch=16, cex=0.6, xlab="Mean log-expression",
- ylab="Variance of log-expression")
- curve(var.fit$trend(x), col="dodgerblue", lwd=2, add=TRUE)
- cur.spike <- isSpike(sce)
- points(var.out$mean[cur.spike], var.out$total[cur.spike], col="red", pch=16)
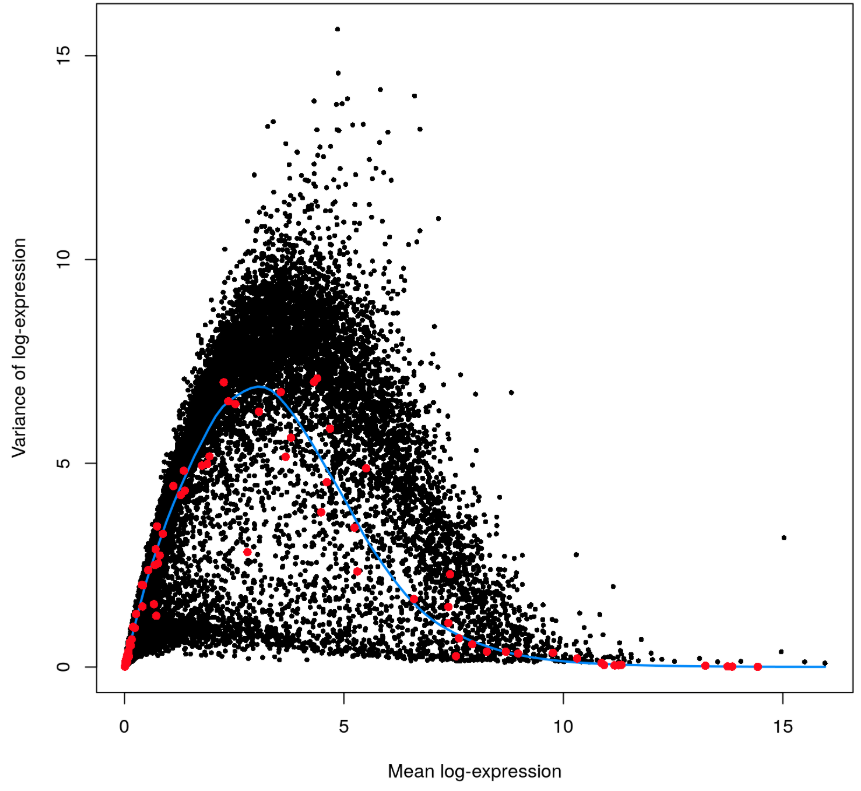
上面decomposeVar()计算的生物因素方差,我们更希望是这样一种情况:某个基因在这一小撮细胞中表达量低,在另外一撮细胞中表达量高。而不是:这个基因在某一两个细胞中表达超高,其他细胞都差不多并且比较低(这种情况实际上依然不能说明基因差异表达,只能说明受离群点的影响)
因此,为了证明上面的结果真的是考虑了所有细胞中基因表达量水平后,得到的生物因素方差结果,选择生物方差前10的基因做个小提琴图,看看到底是由于离群点还是真的是一撮一撮细胞导致的差异
- chosen.genes <- order(var.out$bio, decreasing=TRUE)[1:10]
- plotExpression(sce, features=rownames(var.out)[chosen.genes]) + fontsize
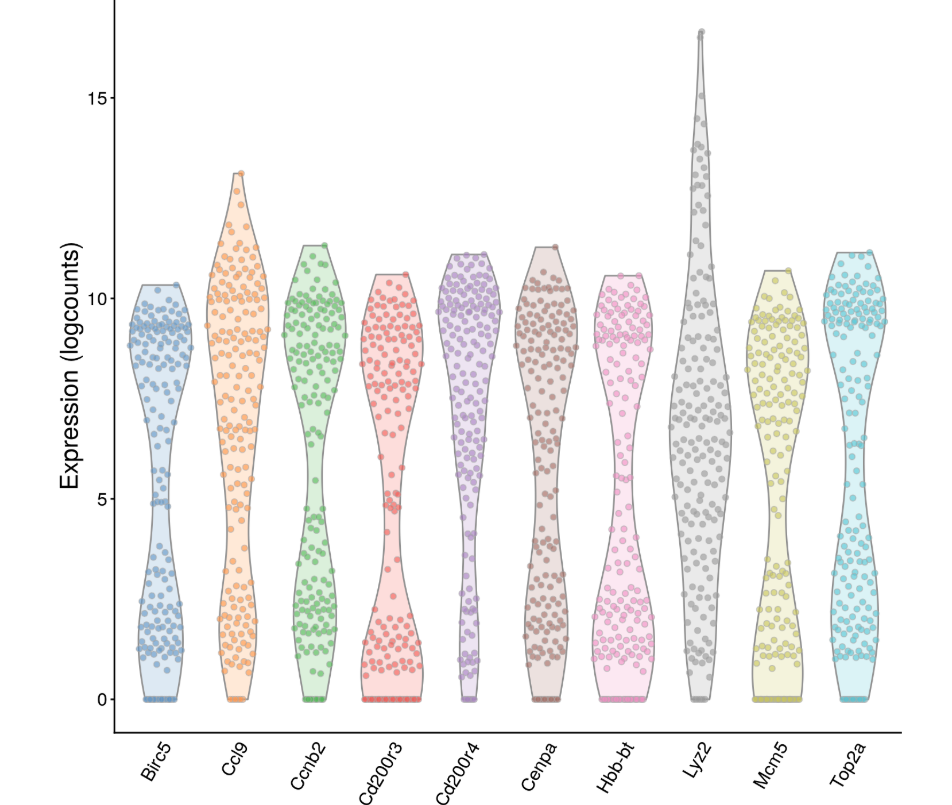
(图中点是一个一个的细胞,纵坐标是这个基因在细胞中的normalized log-expression)
作者建议
- 使用少量的spike-in来拟合技术噪音是很复杂的,另外它们的表达量也并不均一,如果要继续优化这个拟合过程,可以参考:https://bioconductor.org/packages/3.9/simpleSingleCell/vignettes/var.html#details-of-trend-fitting-parameters
- 如果实验中不使用spike-in,可以使用参数
use.spikes=FALSE来拟合,参考:https://bioconductor.org/packages/3.9/workflows/vignettes/simpleSingleCell/inst/doc/var.html#32_when_spike-ins_are_unavailable
9 移除批次效应
前面提到,这个数据包括了两个细胞板信息,在实验过程中,这两个细胞板会存在一些小的不可控的技术差异,这就产生了”批次效应“。这种没有生物学意义的差异我们是不感兴趣的,可以用limma的removeBatchEffect()函数去除。这个函数既可以去掉不感兴趣的因素(在参数batch中指定),又可以保留原来感兴趣的生物学因素(用参数design指定)。
函数的帮助文档对这个参数的定义是这样的:The design matrix is used to describe comparisons between the samples, for example treatment effects, which should not be removed.
- library(limma)
- # -----修改原来的实验比较分组--------
- # 谁在前表示谁作为对照
- sce$Oncogene <- relevel(sce$Oncogene,ref = 'control')
- # -----检查好分组信息后去除批次--------
- assay(sce, "corrected") <- removeBatchEffect(
- logcounts(sce),
- design=model.matrix(~sce$Oncogene),
- batch=sce$Plate
- )
- assayNames(sce)
- ## [1] "counts" "logcounts" "corrected"
去除批次效应对下游的降维、聚类改善效果最为明显。当每个批次中的细胞组成类型已知或者不同批次都是同一种类型细胞只是处理方式不同,这种情况用removeBatchEffect()效果不错;但是大部分的scRNA数据中,每个批次的细胞类型我们可能不会那么确定,不同批次的细胞类型也可能不相同,这种真实复杂的情况下,更推荐使用mnnCorrect()
10 PCA去除表达量中的技术噪音
之前(在第8步)进行了模拟基因表达中的技术噪音,现在就可以利用PCA去除表达量的技术噪音。可以看到,从上到下分析,我们的一个重要目的就是:去掉一切和生物学因素无关的噪音、差异
我们知道PCA目的是降维,那么到底它降低的是什么维度?是什么降低的?
试想,我们得到的单细胞数据一般都有成百上千个细胞样本,这一个细胞都有自己的一个维度,把它们放在一起就是一个高维空间,高维空间中的每一个点就代表一个细胞,点和点之间的离散分布就表示了细胞间全部的差异(包括生物因素差异和技术因素差异)。PCA对这个空间利用坐标轴指定主成分,主成分越靠前,包含的差异成分越丰富。因此我们喜欢使用前两个主成分来做二维PCA图甚至使用三个主成分来做3D PCA plot。
我们认为,技术因素差异和生物因素差异相比,生物差异更能代表大部分的总体差异,而这大部分的总体差异又是体现在前几个PCs。不过,技术因素差异也会单独影响每个基因,虽然它们产生的差异不如生物因素多,但还是有,只不过会体现在后面的主成分中。为了去除这部分技术噪音,可以用denoisePCA() 去除后面几个PCs,那么去掉几个合适?这个函数的算法是:去掉成分中的方差之和 等于 前面模拟结果var.out中全部技术因素的方差之和
- sce <- denoisePCA(sce, technical=var.out, assay.type="corrected")
- dim(reducedDim(sce, "PCA"))
- ## [1] 183 24
最后一共得到了24个主成分,使用这个函数,可以更多地去除其中的技术因素成分,而保留的PCs更多展现生物因素成分差异,可以提高后面聚类的准确性
作者建议
denoisePCA()函数只使用var.out中生物因素方差为正的基因,也就是说,它们的总体技术方差不会高于总体方差- 其中的参数
technical可以直接使用函数var.fit$trend去自行计算,例如下面的代码 - 这里没有对基因丰度进行过滤,是为了保证与一些罕见细胞群体相关的PCs也能被检测到。因为低丰度基因一般在细胞中的方差也比较小,因此它对总体差异的贡献不会太大,它的PC不会太靠前,所以我们不去除它们也并无大碍
- 另外还可以根据过滤后的PCs得到对应的低维度表达矩阵
lowrank,比如下面的代码就是得到了过滤后的24个主成分包含的基因表达矩阵
- sce2 <- denoisePCA(sce, technical=var.fit$trend,
- assay.type="corrected", value="lowrank")
- assayNames(sce2)
- ## [1] "counts" "logcounts" "corrected" "lowrank"
- # 对比一下:这是过滤技术因素主成分以后的表达矩阵
- > assay(sce2, "lowrank")[1:3,1:3]
- SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
- ENSMUSG00000103377 -0.070549323
- ENSMUSG00000103147 0.071665873
- ENSMUSG00000103161 0.007761405
- SLX-9555.N701_S503.C89V9ANXX.s_1.r_1
- ENSMUSG00000103377 -0.030665490
- ENSMUSG00000103147 0.139113948
- ENSMUSG00000103161 0.002681669
- SLX-9555.N701_S504.C89V9ANXX.s_1.r_1
- ENSMUSG00000103377 0.002395618
- ENSMUSG00000103147 0.026324826
- ENSMUSG00000103161 0.002692717
- # 这是过滤技术因素主成分以前的表达矩阵
- > assay(sce2, "corrected")[1:3,1:3]
- SLX-9555.N701_S502.C89V9ANXX.s_1.r_1
- ENSMUSG00000103377 -0.008158873
- ENSMUSG00000103147 0.034282013
- ENSMUSG00000103161 0.005195027
- SLX-9555.N701_S503.C89V9ANXX.s_1.r_1
- ENSMUSG00000103377 -0.008158873
- ENSMUSG00000103147 0.034282013
- ENSMUSG00000103161 0.005195027
- SLX-9555.N701_S504.C89V9ANXX.s_1.r_1
- ENSMUSG00000103377 -0.008158873
- ENSMUSG00000103147 0.034282013
- ENSMUSG00000103161 0.005195027
11 在低维空间可视化
11.1 利用PCA
对前三个主成分进行两两比较,看看细胞之间的联系
具有相似表达谱的细胞距离会更近(可以理解成每个细胞中都是一万多基因表达,这些基因有的表达高有的表达少,这种高低交错的形态就是表达谱。如果两个细胞都有相似的这种高低交错形态,那么它们可能就具有相似的基因表达特性,于是更有可能是同一类细胞。PCA就是衡量每个细胞中这一万多个基因的表达特性,来对这些细胞进行”种群划分“)
首先根据细胞生物学分类(control和treat)画PCA
- plotReducedDim(sce, use_dimred="PCA", ncomponents=3,
- colour_by="Oncogene") + fontsize
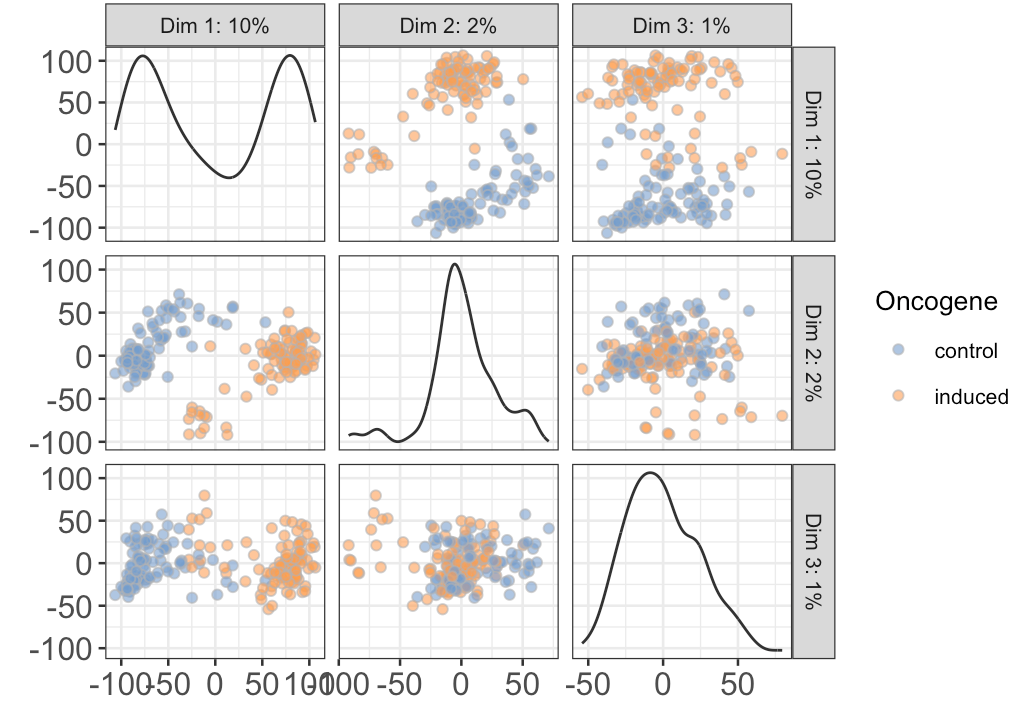
图中可以看到,PC1+PC2的效果是最好的(左侧中部图)
然后根据plate信息画PCA
目的是看看之前的去除批次做的效果如何,如果结果还是和上图一样能分开,那么就表示plate差异依然很大,足够撼动真实的生物学差异
- plotReducedDim(sce, use_dimred="PCA", ncomponents=3,
- colour_by="Plate") + fontsize
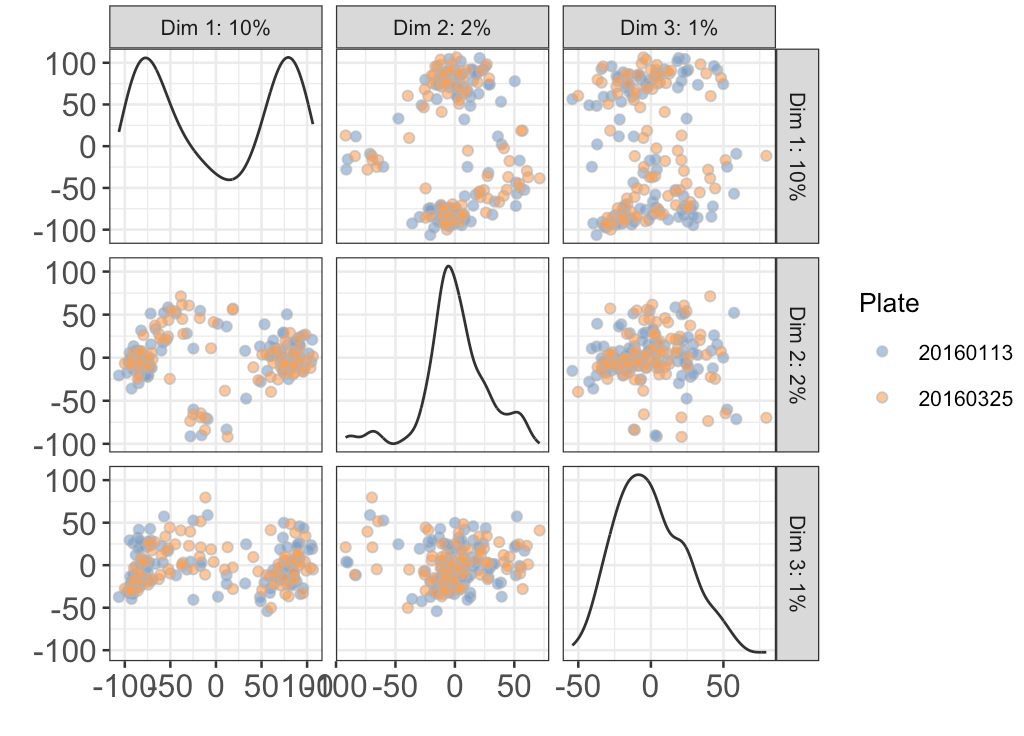
还是看最具代表性的PC1+PC2:不同plate的细胞分不开,说明之前removeBatchEffect()是成功的
注意:
plotReducedDim()会使用之前利用denoisePCA()得到的结果,而且可以一次画多个主成分两两比较,视觉效果更直观。
作者建议
- 每个作图函数,都能根据细胞类型去调整点的颜色和大小,一般会设置:
size_by=和shape_by= - 虽然还有许多主成分,但它们包含的信息量远不如前几个主成分
11.2 利用tSNE
另一个常用的降维算法就是t-stochastic neighbour embedding (t-SNE),是基于在邻域图上随机游走的概率分布。它比PCA的强大之处在于,如果细胞群体很多,tSNE依然可以很好地将细胞按一定规则区分。这是因为tSNE能在高维空间中直接捕获细胞非线性关系,而PCA必须在线性的坐标轴上去展示它们。不过tSNE的计算量会更大,而且参数设置更复杂,一方面针对tSNE每次映射的坐标都不同,要设置随机种子,方便别人重复;另一方面需要设置perplexity参数,它会影响最后显示的细胞分类情况,perplexity的含义是:the number of close neighbors each point has
下面会利用PCA降维结果继续使用多个perplexity值进行tSNE可视化
- set.seed(100)
- out5 <- plotTSNE(sce, run_args=list(use_dimred="PCA", perplexity=5),
- colour_by="Oncogene") + fontsize + ggtitle("Perplexity = 5")
- set.seed(100)
- out10 <- plotTSNE(sce, run_args=list(use_dimred="PCA", perplexity=10),
- colour_by="Oncogene") + fontsize + ggtitle("Perplexity = 10")
- set.seed(100)
- out20 <- plotTSNE(sce, run_args=list(use_dimred="PCA", perplexity=20),
- colour_by="Oncogene") + fontsize + ggtitle("Perplexity = 20")
- multiplot(out5, out10, out20, cols=3)
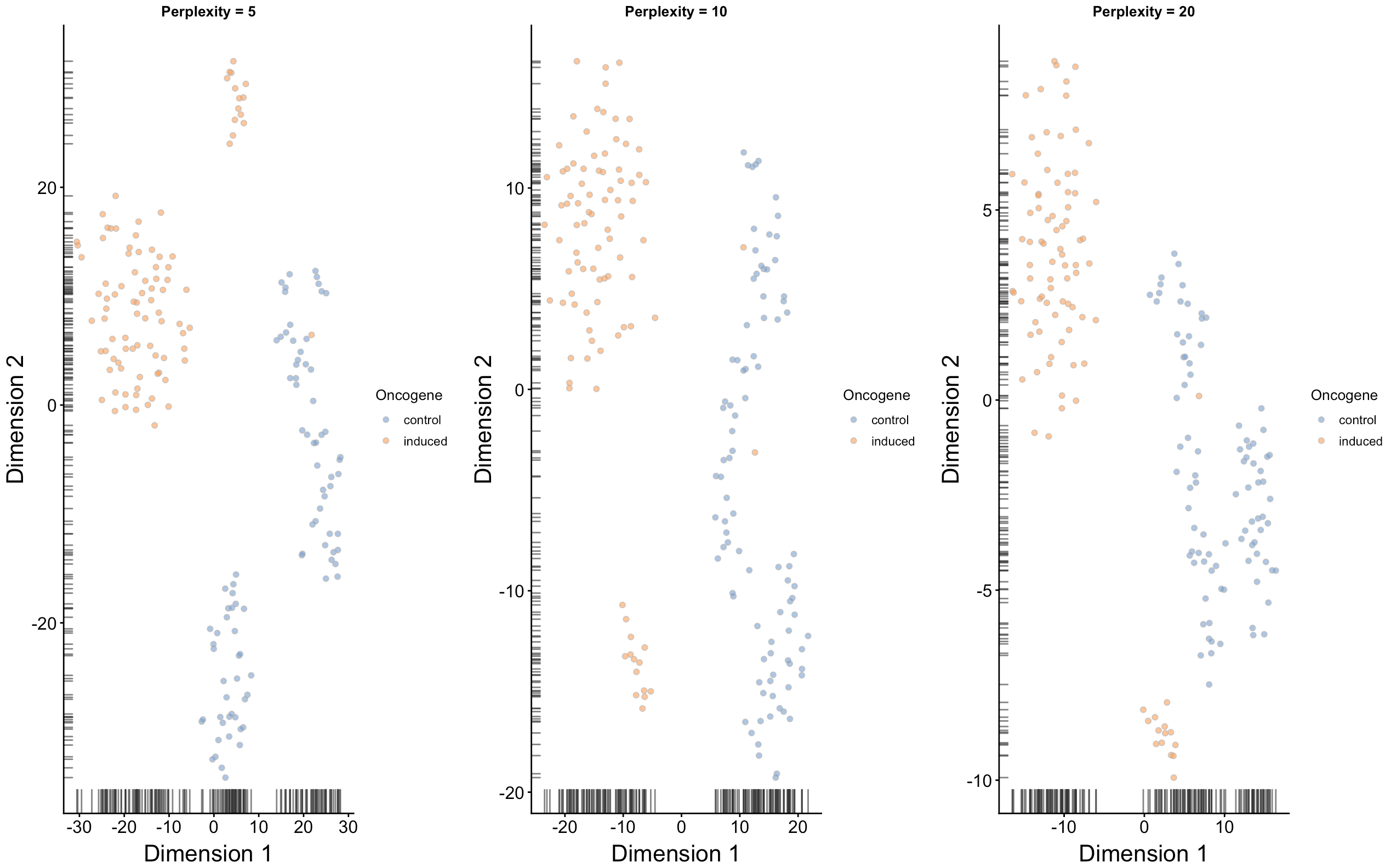
因为tSNE是一个随机的算法,所以我们要多试验几遍,确保结果具有代表性(相当于多做几次实验重复,增加可靠性);最终的脚本要使用set.seed()保证可重复性;不同的perplexity 会影响细胞在低维空间的分布
从上面图中,如果认为perplexity=20效果还不错,因此下面继续跑runTSNE时就可以设置perplexity=20
- set.seed(100)
- sce <- runTSNE(sce, use_dimred="PCA", perplexity=20)
- reducedDimNames(sce)
- ## [1] "PCA" "TSNE"
除此以外,还有许多降维的方法:例如multidimensional scaling、 diffusion maps,当然不同算法各有自己的适用范围,比如 diffusion maps(plotDiffusionMap) 会将细胞放在一个连续的轨迹上,这在推断分化过程方面是很有效的
作者建议
- 如何解释tSNE图?可以参考https://distill.pub/2016/misread-tsne/ 其中映射到二维坐标上的cluster之间的距离其实没什么意义,还有cluster的大小(也就是其中点的分布稀疏程度,而不是指其中包含点的多少。对于同等数量的点,分布分散一点形成的cluster就大)也是没什么具体意义。它只是一种二维空间再现高维空间的手段
perplexity的值一般设为5-50,可以看一下上面链接中给出的测试图:当这个值不在5-50范围时,点聚成的形状就很奇怪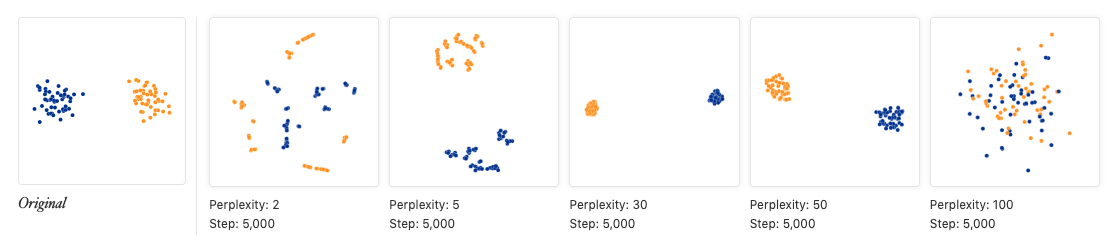
- 另外画tsne图时一般要重复很多次,因为重复次数越多,画出的图形状越稳定,上图中使用了5000次重复,下图如果仅仅使用120次重复,结果会是:
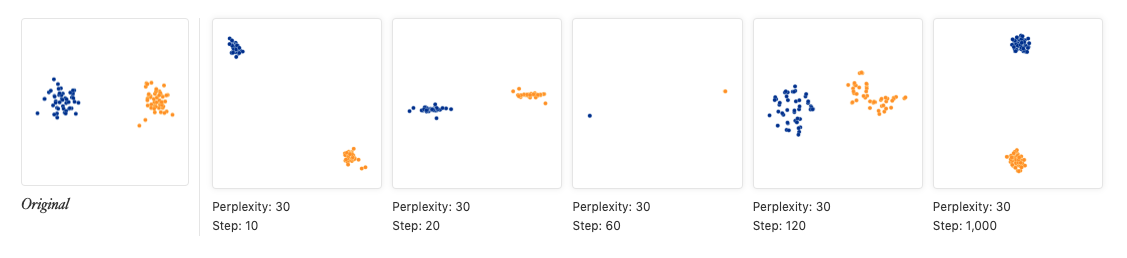
- tsne图中的点距离并没有意义,用两张图就能说明:
第一张:本来绿色的点和其他两种是有距离的,但是当设置的perplexity增大发现,图中它们距离是差不多相等的。Wait!那如果设成50不就可以了吗?
从这个图中看好像是的,但是...
如果增加原本点的个数,依然是绿色与其他两种有原始距离,使用perplexity=50,结果依然得不出相似的结论,说明perplexity不是一个固定的值,它会根据分析点的数量而变化,得到的结果也不能反应原始数据结构

- tSNE能做的,只是将原本聚在一起的,现在依旧聚在一起,并且有时会给你一些数据形状
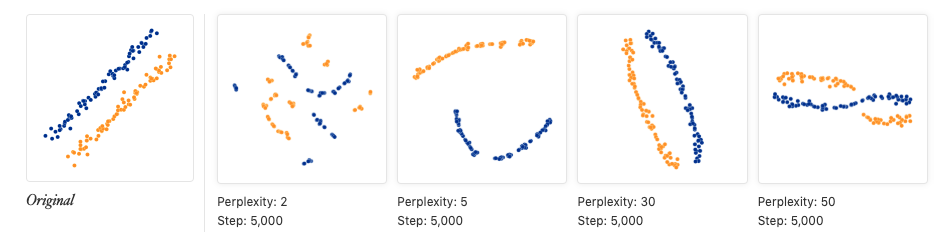
- tSNE使用很流行的一个原因就是:它很灵活,经常能帮助寻找到数据的结构(像线性方法PCA就不可以),不过想要找点的距离远近就不要看tSNE了
12 自己进行细胞聚类
12.1 降维结果+层次聚类=》分群
目标:利用去除技术噪音的PCA结果进行细胞聚类,推断细胞亚群。
前面去除技术噪音中使用了denoisePCA函数,最后保留了24个主成分,存储在reducedDim(sce, "PCA")中,其中行是细胞,列是主成分(从PC1到PC24)。
思路就是:先提取PCA结果中细胞与主成分PC1、PC2的坐标,然后根据细胞在空间上的欧式距离进行层次聚类(对行进行操作,也就是这里的细胞),并且使用Ward标准去降低每个cluster内的总体方差,保证了每个cluster中的细胞都含有相似的基因表达模式
- # 三步走:取PCA结果=》计算细胞欧式距离=》降低cluster内差异
- pcs <- reducedDim(sce, "PCA")
- my.dist <- dist(pcs)
- my.tree <- hclust(my.dist, method="ward.D2")
接着利用dynamic tree cut的方法得到cluster的数量,在复杂的聚类数中相比cutree更有优势 (Langfelder, Zhang, and Horvath 2008) 。但是函数计算的毕竟是统计角度的cluster,我们还需要人工干预帮忙校正一下。比如设置:cutHeight、minClusterSize 。
- library(dynamicTreeCut)
- my.clusters <- unname(cutreeDynamic(my.tree, distM=as.matrix(my.dist),
- minClusterSize=10, verbose=0))
- # minClusterSize默认值是20,这里设置小一点是避免一些偏远的小群体错误地凑在一起
然后,利用得到的细胞分群结果与已知的分类因子进行结合,首先看得cluster与批次之间的关系:
- table(my.clusters, sce$Plate)
- ##
- ## my.clusters 20160113 20160325
- ## 1 41 39
- ## 2 19 20
- ## 3 16 11
- ## 4 10 14
- ## 5 5 8
发现其中每个cluster基本都包含两个批次,并且数量都差不多,表明这里的聚类结果不受批次效应影响。
再看cluster与生物差异之间的关系:
- table(my.clusters, sce$Oncogene)
- ##
- ## my.clusters induced control
- ## 1 80 0
- ## 2 0 39
- ## 3 0 27
- ## 4 0 24
- ## 5 13 0
发现其中的cluster在处理和对照之间存在很大差异,更加验证了是由于生物因素导致的分群
得到cluster后,用tsne结果进行映射
- sce$cluster <- factor(my.clusters)
- plotTSNE(sce, colour_by="cluster") + fontsize
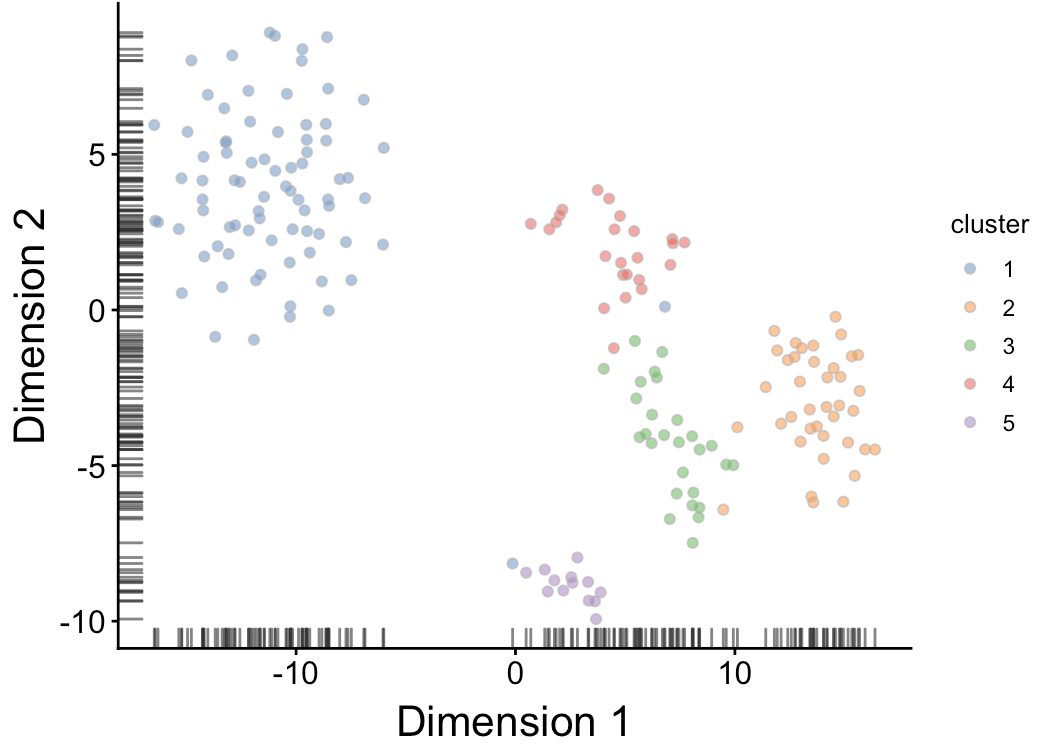
检查分群结果
一般来说,分群结果分的越开,说明效果越好。如果感觉用肉眼观察tsne图不容易判断的话,还有一种方法可以帮助我们判断,学名叫”silhouette width“(轮廓图)。先上代码和图:
- library(cluster)
- clust.col <- scater:::.get_palette("tableau10medium") # hidden scater colours
- sil <- silhouette(my.clusters, dist = my.dist)
- sil.cols <- clust.col[ifelse(sil[,3] > 0, sil[,1], sil[,2])]
- sil.cols <- sil.cols[order(-sil[,1], sil[,3])]
- plot(sil, main = paste(length(unique(my.clusters)), "clusters"),
- border=sil.cols, col=sil.cols, do.col.sort=FALSE)
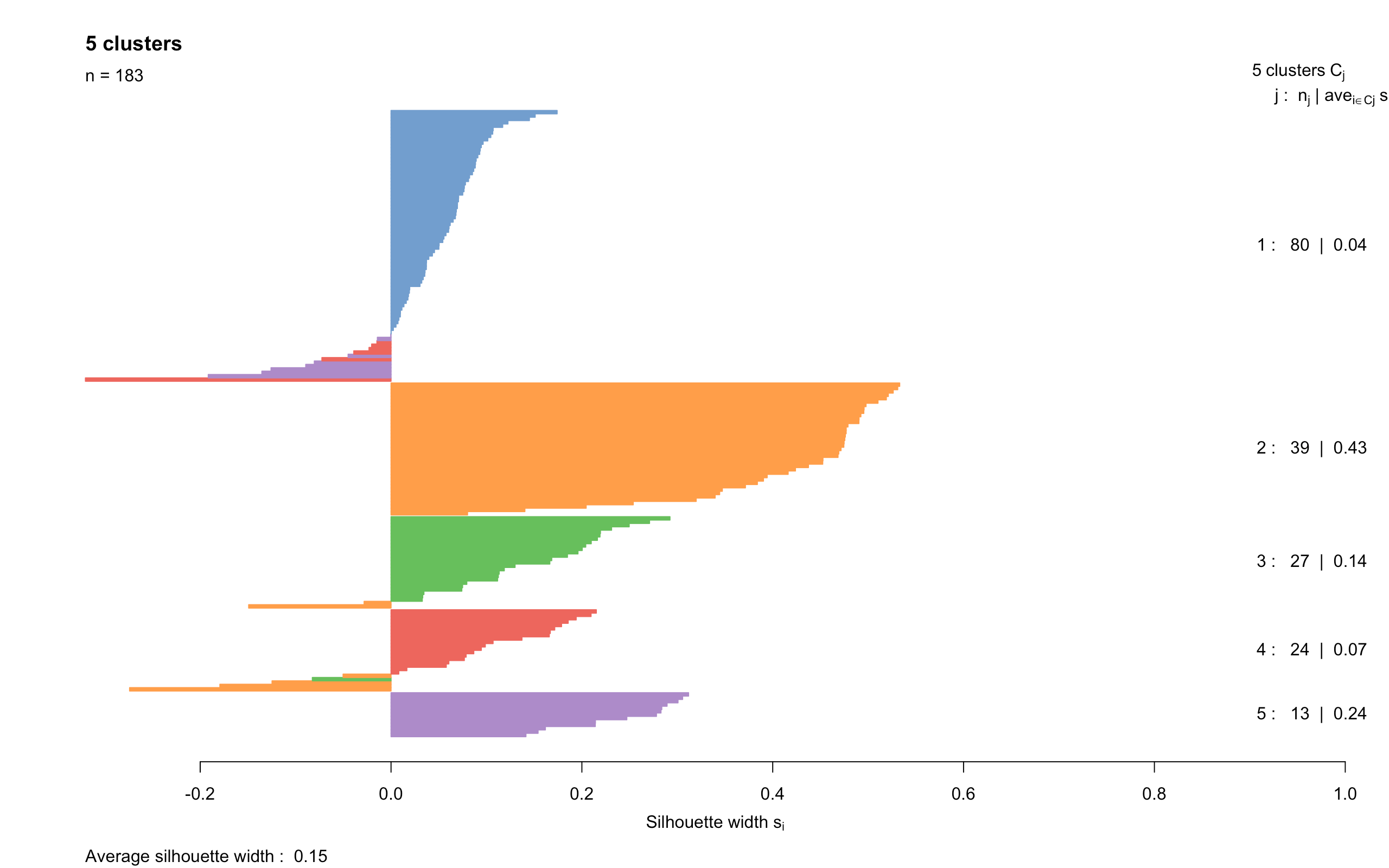
- 这个图是对上面得到的各个cluster中的细胞做的barplot,每个cluster是一种颜色。
- 如果一个细胞的横坐标width值为正并且它越大,表示相比于其他clusters的细胞,这个细胞和它所在cluster中的其他细胞更接近;如果width为负,就表示这个cluster的这个细胞和其他cluster的细胞更接近,也就是说分群效果不好
- 利用这个图,我们就能调整之前的参数,来调整分群效果,不过也不需要太过纠结完美的分群结果
- 从图中可以看到,大多数细胞的width数值比较小,表明cluster之间的分离效果一般。很有可能是”过度分群“,比如原来都属于treat组的cluster又继续分成了小cluster,那么它们之间分离效果肯定要差。
作者建议
- 聚类也可以用
quickCluster函数,关于它的利弊:more robust to noise and normalization errors, but is also less sensitive to subtle changes in the expression profiles
12.2 每个cluster检测marker基因
得到了不同的cluster,就可以利用
findMarkers()去为每个cluster寻找marker基因。
它的方法是:两两cluster之间对每个基因的log表达量进行 Welch t-tests (Soneson and Robinson 2018)。这样就能找到每个cluster中的差异表达基因(前提是掩盖掉非生物因素,例如批次信息)。得到的top 差异基因能更好地作为marker基因候选,原因就是:既然它们是最具有差异代表性的,那么利用它们就能区分不同cluster的细胞,也就实现了marker的目的。【当然,还有许多细胞类型自己特有的基因,也是作为marker的候选者】
- markers <- findMarkers(sce, my.clusters, block=sce$Plate)
每个cluster与其他cluster的两两比较结果会整合到每个cluster的差异基因结果数据框中
例如:看一下第1个cluster的top10差异基因
- marker.set <- markers[["1"]]
- head(marker.set, 10)
- ## DataFrame with 10 rows and 7 columns
- ## Top p.value FDR logFC.2
- ## <integer> <numeric> <numeric> <numeric>
- ## Aurkb 1 6.65863261824492e-75 1.58362259559721e-70 -7.37163350569914
- ## Tk1 1 6.41442387689833e-64 3.81385607660686e-60 -4.92754579848056
- ## Myh11 1 4.95086465211539e-49 9.81220116843835e-46 4.42159318376489
- ## Cdca8 1 2.22334940769686e-46 3.5251945975503e-43 -6.84273526334783
- ## Ccna2 2 1.16841222330534e-68 1.38941739534356e-64 -7.3079756458424
- ## Rrm2 2 1.48873842020081e-56 5.05809512109088e-53 -5.52120322191947
- ## Cks1b 2 3.83636139889977e-39 2.40105745131667e-36 -6.6792118199827
- ## Pirb 2 1.83893803490065e-34 6.15992440620318e-32 5.25803749166673
- ## Pimreg 3 7.41004737119548e-68 5.87443855430467e-64 -7.30454210816126
- ## Pclaf 3 8.9651722100101e-51 2.13218690670669e-47 -5.60087985621813
- ## logFC.3 logFC.4 logFC.5
- ## <numeric> <numeric> <numeric>
- ## Aurkb -6.72799345321135 -1.95039440976238 -6.91128802096519
- ## Tk1 -7.74065113926215 -3.53749565362853 -4.63516273649457
- ## Myh11 4.30812918035861 4.45235717737968 1.0413149433198
- ## Cdca8 -4.88595092732349 -2.43821402084038 -7.12791471326961
- ## Ccna2 -6.9676852052539 -2.46589325823089 -7.12692721843331
- ## Rrm2 -7.94685699818751 -3.19173143688883 -5.42878091964762
- ## Cks1b -5.92137181826573 -4.37146346518812 -6.214592473138
- ## Pirb 5.18195596569259 5.87631057587633 0.0704964218555272
- ## Pimreg -5.91099762335684 -0.874660676141792 -7.01798853404623
- ## Pclaf -7.56997893312346 -2.36631043435985 -5.16956927698937
可以将全部cluster的marker基因保存起来
- write.table(marker.set, file="416B_marker_1.tsv", sep="t",
- quote=FALSE, col.names=NA)
最后热图对cluster1的marker基因可视化
- top.markers <- rownames(marker.set)[marker.set$Top <= 10]
- plotHeatmap(sce, features=top.markers, columns=order(sce$cluster),
- colour_columns_by=c("cluster", "Plate", "Oncogene"),
- cluster_cols=FALSE, center=TRUE, symmetric=TRUE,
- zlim=c(-5, 5),
- show_colnames = F)
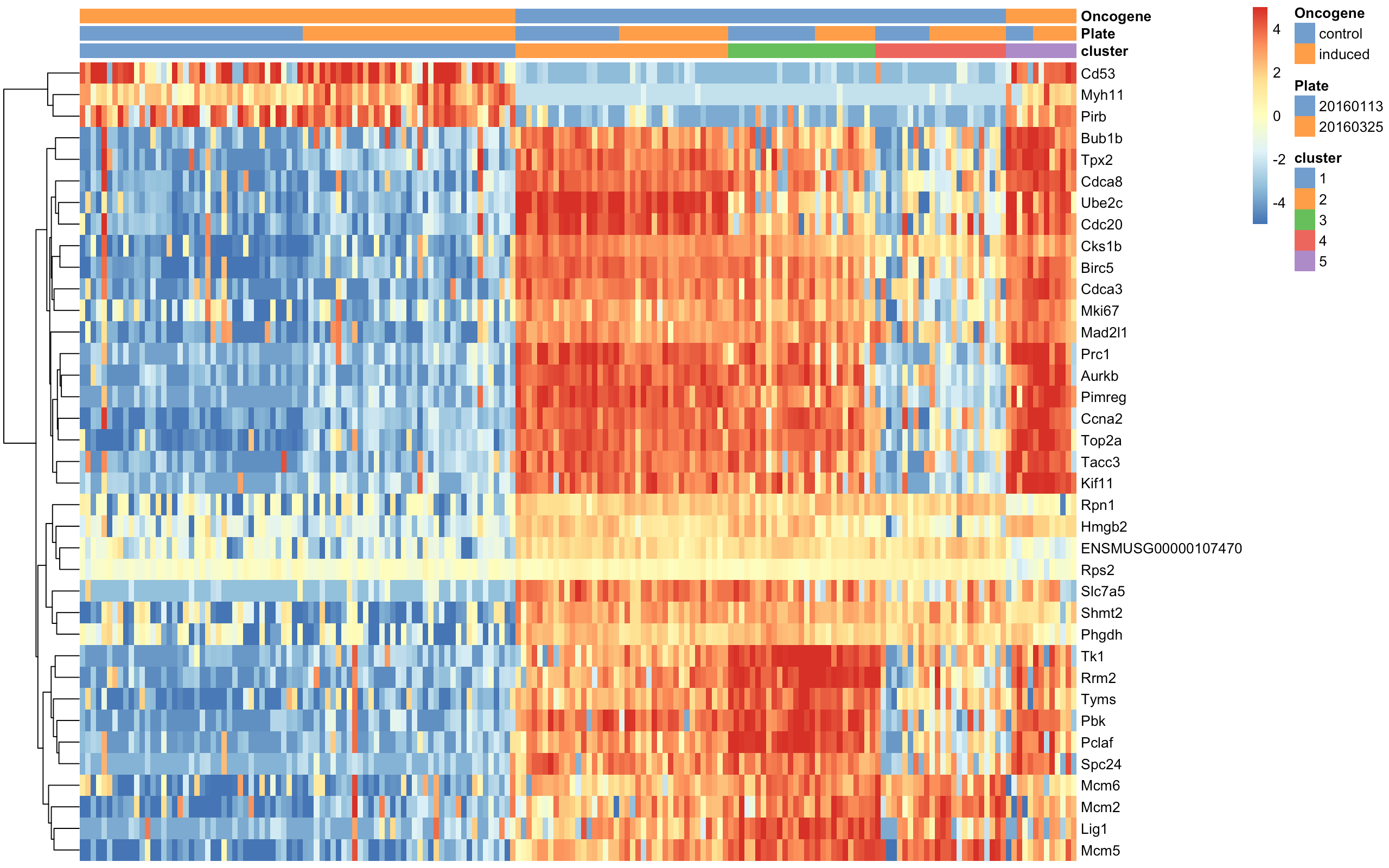
从图中粗略地看一看:cluster1包含了处理组(oncogene-induced)细胞,另外第5群也是处理组细胞。如果生物背景足够的话,也能推断出这些细胞与某些DE基因之间的联系。
「需要注意的地方」
当然,不是所有的marker基因都在所选cluster都是上调或下调的,它不仅仅是差异基因这么简单。如果只选差异基因,这样会过滤掉很多重要的marker基因。
例如,在CD4+-only, CD8+-only, double-positive and double-negative T cells混合的细胞群体中,重要的细胞marker基因Cd4或Cd8 都不会被检测到,因为这两个基因会在其中两个类型的细胞群体中都存在(比如Cd4 会存在于CD4+-only和 double-positive两种细胞类型中)
这里强烈建议挑选一些已经验证过的细胞marker基因,比如
- CellMarker数据库(http://biocc.hrbmu.edu.cn/CellMarker/):整理了100,000+发表的文献,包含人的158个组织 (亚组织)的467个细胞类型的13,605个Marker基因,和鼠的81个组织 (亚组织)的389个细胞类型的9, 148个Marker基因
- PanglaoDB(https://panglaodb.se/):小鼠的170种组织954个样本近400W细胞和来自人的68种组织279个样本100w+细胞
- 小鼠Mouse Cell Atlas(http://bis.zju.edu.cn/MCA/)
- signatureDB(https://pldaniels.com/signaturedb/)
最好再加上一些定量实验,比如fluorescent in situ hybridisation or quantitative PCR,确定细胞亚群真实存在,而不仅仅是因为我们分析得到是这样
作者建议
- 如果只想提取cluster中的上调表达基因,就可以在
findMarkers中指定参数direction="up"。应用在高度异质性群体中,可以更快速判断不同的cluster findMarkers除了将一个cluster与其他各个cluster两两比较之外,还能对某个cluster和其他全部clusters进行差异分析,设置参数pval.type="all";如果再和direction="up"连用,就可以用来鉴定每个cluster中特异的marker基因。但是如果存在”过度聚类“的情况,原本一个cluster分成了两个,那么这个特异的marker基因就不会存在。- 另外,这里的p值不能简单理解为显著性。具体原因在:
https://bioconductor.org/packages/3.9/simpleSingleCell/vignettes/de.html#misinterpretation-of-de-$p$-values
结语
基础分析到此结束
那么高级分析都有哪些呢?这里举一些例子:
- pseudotime拟时序分析(比如探索分化过程): monocle (Trapnell et al. 2014) or TSCAN (Ji and Ji 2016)
- cell-state hierarchies:sincell (Julia, Telenti, and Rausell 2015)
- oscillatory behaviour:Oscope (Leng et al. 2015)
- 高变化基因(HVGs)的GSEA分析:结合异质性鉴定生物学通路或代谢途径:scde (Fan et al. 2016)
心得体会
理解这长长的英文文档不是件容易事,但是作者写的很简单易懂,辅助之前的知识,再一次添加了一些知识。