

1 低质量细胞的影响
- 细胞破坏后,可能会导致线粒体或核RNAs占比升高(就是上面解释的大量细胞质中mRNA流失,而线粒体或核RNAs含量基本不变),很有可能会根据这个结果形成自己的一个个cluster
- 低质量的细胞一般文库比较小,而差异分析之前一般对文库大小进行一个归一化。比如正常细胞文库大小是100,某个基因表达量是2;损伤细胞的文库大小是10,这个基因表达量还是2。归一化后,损伤细胞中的这个基因表达量计算结果明显会高于正常细胞,呈现一种“本来不优秀,但班里人少了,排名就上升”的状态
- 细胞损伤可能会伴随RNA的流失,因此许多基因可能会被认为“下调”,尤其体现在细胞质核糖体RNA(另外还包括一些细胞质转录本)
- 影响方差估计和PCA结果。真实情况下,可能一个基因在两个细胞中差异并不显著,但是由于其中一个细胞质量低,导致基因表达量在这两个细胞中差异明显;反映在PCA结果就是:前几个主成分会抓取细胞质量的差异,因为这种差异体现得更明显,而将真正的生物学因素放到了后面几个主成分中,因此得到的PCA结果其实也只是反映了细胞质量的差异,而非真正的生物学差异
2 需要注意的点
- 如果一个细胞群体异质性较高,那么很有可能一些高质量细胞本身表达的数量就是比其他细胞少,但事实上它不是技术误差造成的。因此不能通过一个固定的阈值进行过滤,而要“因地制宜”,根据每群细胞各自的特性(比如各自的中位值),然后结合一定的统计指标(例如3倍的MAD)
- 过滤的细胞会不会属于某一个具有生物意义的细胞类群,如果真的是,那么就会有相应的marker基因高表达。
3 初步看一下
先做一个散点图,重点看看有没有基因明显向舍弃细胞方向偏移:
- # 计算丢弃和保留的细胞平均表达量(这里的平均表达量不是我们直接用apply+mean得出的结果,它计算了size factor
- library(SingleCellExperiment)
- sce.full.416b <- readRDS("416B_preQC.rds")
- library(scater)
- lost <- calcAverage(counts(sce.full.416b)[,!sce.full.416b$PassQC])
- kept <- calcAverage(counts(sce.full.416b)[,sce.full.416b$PassQC])
- # 在上面得到的平均值中,将每个数都与平均值中(除0以外)最小的数进行比较,取最大的那个值作为最终的平均值
- capped.lost <- pmax(lost, min(lost[lost>0]))
- capped.kept <- pmax(kept, min(kept[kept>0]))
- plot(capped.lost, capped.kept, xlab="Average count (discarded)",
- ylab="Average count (retained)", log="xy", pch=16)
- is.spike <- isSpike(sce.full.416b)
- points(capped.lost[is.spike], capped.kept[is.spike], col="red", pch=16)
- is.mito <- rowData(sce.full.416b)$is_feature_control_Mt
- points(capped.lost[is.mito], capped.kept[is.mito], col="dodgerblue", pch=16)
calcAverage具体操作如下:The size-adjusted average count is defined by dividing each count by the size factor and taking the average across cells. All sizes factors are scaled so that the mean is 1 across all cells, to ensure that the averages are interpretable on the scale of the raw counts.
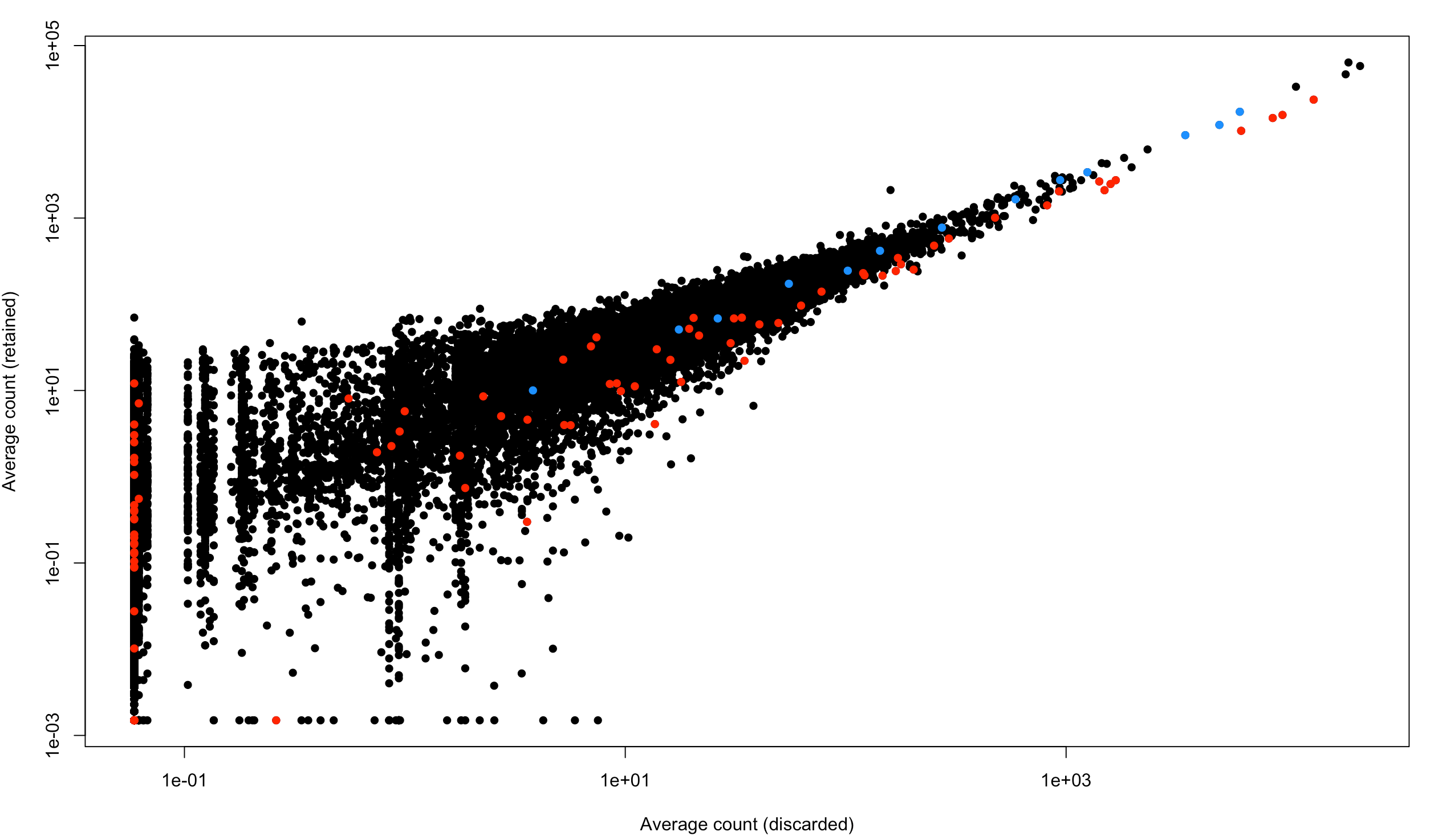
图中表示了舍弃的细胞和保留的细胞中基因平均表达量。每个点是一个基因,红色是spike-in,蓝色是线粒体基因
看到:整体方向还是偏向于y轴,虽然也有个别的点是偏向右下方的,下面👇就看看最偏右下方的那些点到底是不是marker基因?
4 认真再检查一下
方法就是:计算retain和discard两组细胞的基因表达量差异倍数( log-fold changes)。
核心就是:利用了edgeR的predFC函数,因为我们并不关心这两组之间的差异,这是想看看哪些基因在discard组更高表达一些。于是用这个函数分别根据每个组的表达量,再设置一个design实验设计矩阵就可以。没有正常做差异分析那么严格
- library(edgeR)
- #!! 重点在于DGEList中group的设置:数小的是control,也就是logFC的分母(自己也可以将group调换一下试试,结果看看是不是相反的)
- y <- cbind(lost, kept)
- y <- DGEList(y, group=c(2,1))
- design <- model.matrix(~group, data=y$samples)
- predlfc <- predFC(y, design)[,2]
- info <- data.frame(logFC=predlfc, Lost=lost, Kept=kept,
- row.names=rownames(sce.full.416b))
- head(info[order(info$logFC, decreasing=TRUE),], 20)
- # edgeR实验设计矩阵长这样:
- > design
- (Intercept) group2
- lost 1 1
- kept 1 0
- attr(,"assign")
- [1] 0 1
- attr(,"contrasts")
- attr(,"contrasts")$group
- [1] "contr.treatment"
结果就得到了:
- ## logFC Lost Kept
- ## ENSMUSG00000104647 6.844237 7.515034 0.000000000
- ## Nmur1 6.500909 5.909533 0.000000000
- ## Retn 6.250501 10.333172 0.196931018
- ## Fut9 6.096356 4.696897 0.010132032
- ## ENSMUSG00000102352 6.077614 9.393793 0.206637368
- ## ENSMUSG00000102379 6.029758 4.244690 0.000000000
- ## 1700101I11Rik 5.828821 4.483094 0.039199404
- ## Gm4952 5.698380 6.580862 0.172999108
- ## ENSMUSG00000106680 5.670156 3.389236 0.005234611
- ## ENSMUSG00000107955 5.554616 5.268508 0.132532601
- ## Gramd1c 5.446975 4.435342 0.103783669
- ## Jph3 5.361082 4.696897 0.139188080
- ## ENSMUSG00000092418 5.324462 3.395752 0.056931488
- ## 1700029I15Rik 5.316226 8.199510 0.394588776
- ## Pih1h3b 5.307439 2.546814 0.000000000
- ## ENSMUSG00000097176 5.275459 2.541927 0.003772842
- ## Olfr456 5.093383 2.186536 0.000000000
- ## ENSMUSG00000103731 5.017303 3.315016 0.107148909
- ## Klhdc8b 4.933215 19.861036 1.635081878
- ## ENSMUSG00000082449 4.881422 1.878759 0.000000000
挑出来top20在discard中相对高表达的基因,但其中并没有marker基因,这就足够了。不用关心logFC的实际数值,眼前的高可能是因为细胞破损后,计算得到的线粒体基因、细胞质核糖体基因或者核RNA占比升高,计算出来的的表达量也升高,因而比正常的细胞要高,不过这是一种“虚假繁荣”的假象而已。
5 如何避免丢失一些细胞类型
最直接的方法就是修改(上面4.2中isOutlier函数的nmads=参数) ,另外如果我们知道哪些细胞属于什么细胞类型,也可以提前定义出来避免过滤掉。最粗暴的还是不进行任何的细胞过滤,这样虽然确保了不会丢失,但同时增加了无用细胞的占比。因此,要不要过滤、怎么过滤还是要取决于个人的安排。
不得不说,得到的细胞质量和细胞类型还真有一定的关系。如果某种细胞就是在细胞提取阶段不配合,导致后来检测到这群细胞都有损伤,最好还是在保证实验设计的前提下去掉它们=》“牺牲小我保存大我”
6 其他一些质控方法
- 使用固定的阈值:比如设置文库大小阈值为100000,表达基因数量4000。但是这些都需要具有实际的经验,最好还是不要乱设置这些值,因为很有可能会由于自己的随意发挥,而抛弃了一些珍贵的细胞类型
- 看基于质控结果进行PCA分析的离群点: 前期会对每个细胞统计总reads数、总基因数等6个指标,然后用这些指标进行PCA。具体
runPCA的设置如下图:
如果PCA的结果有离群点,就说明在这些指标中(不论太大或太小)某些细胞是“脱离群众的”。举个例子:
- sce.tmp <- runPCA(sce.full.416b, use_coldata=TRUE, detect_outliers=TRUE)
- ##
- ## FALSE TRUE
- ## 187 5
- 另外还可以参考2019年出的cellity包
7 一个“太随意”的错误案例
例如:在没有任何先验知识情况下,我们直接对一个单细胞对象中一个统计指标(total_counts)设置一个阈值(1000)进行过滤:
- wrong.keep <- sce.pbmc$total_counts >= 1000
- lost <- calcAverage(counts(sce.pbmc)[,!wrong.keep])
- kept <- calcAverage(counts(sce.pbmc)[,wrong.keep])
- # 然后同上面一样做一个散点图
- # Avoid loss of points where either average is zero.
- capped.lost <- pmax(lost, min(lost[lost>0]))
- capped.kept <- pmax(kept, min(kept[kept>0]))
- plot(capped.lost, capped.kept, xlab="Average count (discarded)",
- ylab="Average count (retained)", log="xy", pch=16)
- platelet <- c("PF4", "PPBP", "SDPR")
- points(capped.lost[platelet], capped.kept[platelet], col="orange", pch=16)
很明显,这个图有点向下偏,也就是说有一些基因在discard组中表达量更多。然后测试了3个血小板相关的基因(橘黄色),发现它们确实在discard组中表达量更高,说明其中有部分血小板细胞被丢掉了
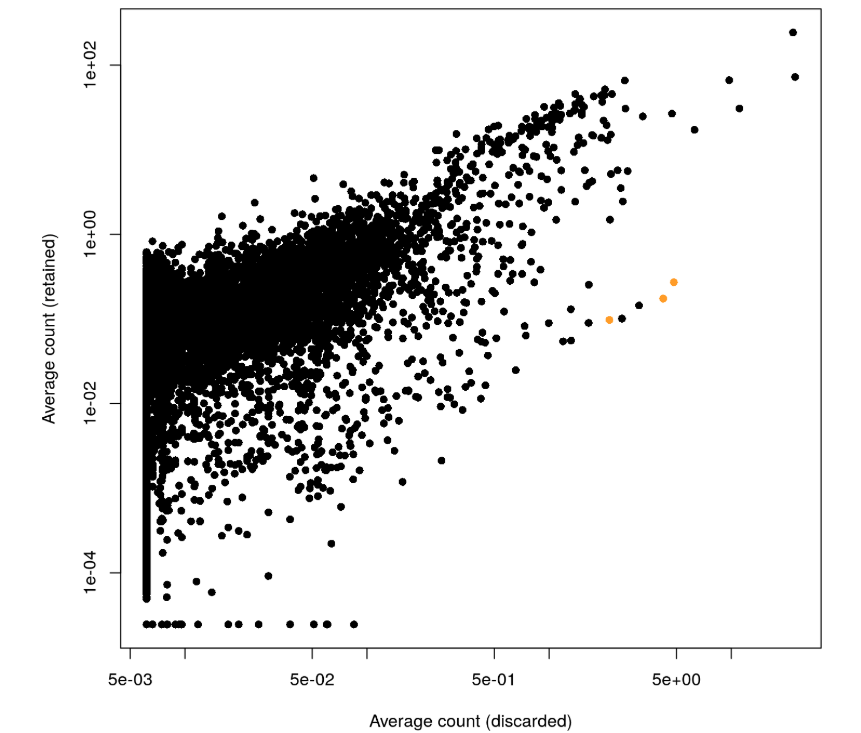
接着用logFC再检验一下:
- coefs <- predFC(cbind(lost, kept), design=cbind(1, c(1, 0)))[,2]
- info <- data.frame(logFC=coefs, Lost=lost, Kept=kept,
- row.names=rownames(sce.pbmc))
- head(info[order(info$logFC, decreasing=TRUE),], 20)
- ## logFC Lost Kept
- ## PF4 6.615671 4.2289086 0.17421441
- ## PPBP 6.495522 4.8409530 0.27144834
- ## HIST1H2AC 6.291680 3.1195253 0.14435051
- ## GNG11 6.162314 2.5132359 0.10103056
- ## SDPR 5.950336 2.1430640 0.09754273
- ## TUBB1 5.610679 1.6478079 0.08989034
- ## CLU 5.462238 1.3202347 0.05559122
- ## ACRBP 5.325560 1.1933566 0.05436568
- ## NRGN 5.118699 1.3155255 0.12992550
- ## RGS18 5.009974 1.6512487 0.25377562
- ## MAP3K7CL 4.898185 0.9950268 0.08957073
- ## SPARC 4.646432 0.6559761 0.02487467
- ## MMD 4.616159 0.7450948 0.06367575
- ## PGRMC1 4.503524 0.7335820 0.08248177
- ## CMTM5 4.166574 0.4469937 0.01643304
- ## TSC22D1 4.120268 0.5128047 0.05920195
- ## HRAT92 4.116163 0.4214188 0.01144488
- ## GP9 4.091146 0.4610813 0.03703803
- ## CTSA 3.986470 0.8294337 0.27097756
- ## MARCH2 3.966528 0.5655020 0.12228759
发现,血小板几个相关基因的确在discard组中更高,说明之前按照sce.pbmc$total_counts >= 1000的过滤是错误的。而不检查有没有先验知识的话,后面分群我们也得不到这部分细胞