将SRA转为fastq
我们使用fastq-dump这款软件,它是sra-tool中的一个工具,使用conda安装即可
- conda install -c bioconda sra-tools
关于这个软件,参数设置一般比较简单(虽然帮助文档写的不是那么好理解)
- wkd=/home/project/single-cell/MCC
- cd $wkd/raw/P2586-4
- cat SRR_Acc_List-2586-4.txt |while read i
- do
- time fastq-dump --gzip --split-3 -A $i ${i}.sra && echo "** ${i}.sra to fastq done **"
- done
其中主要使用了三个参数:
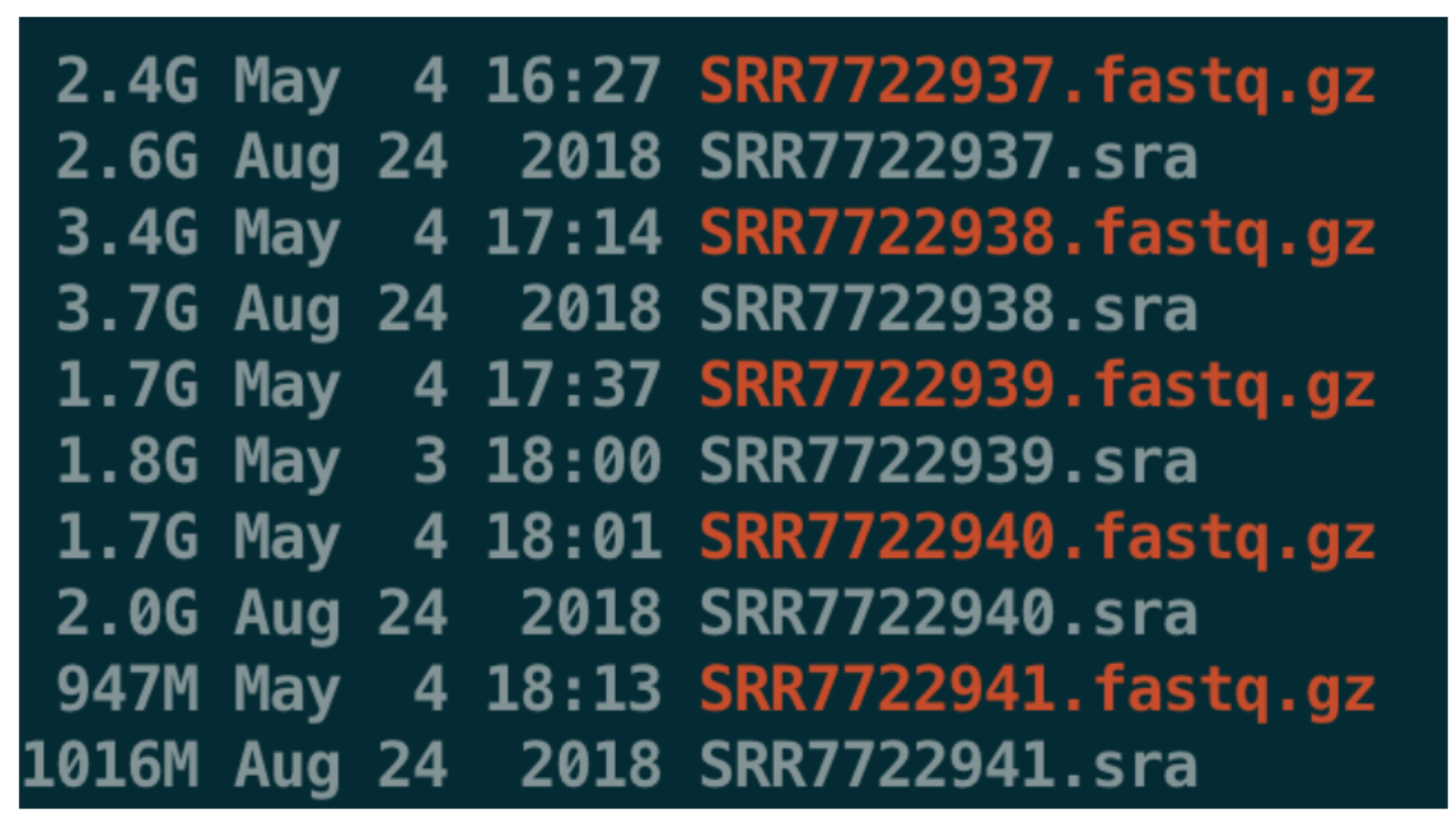
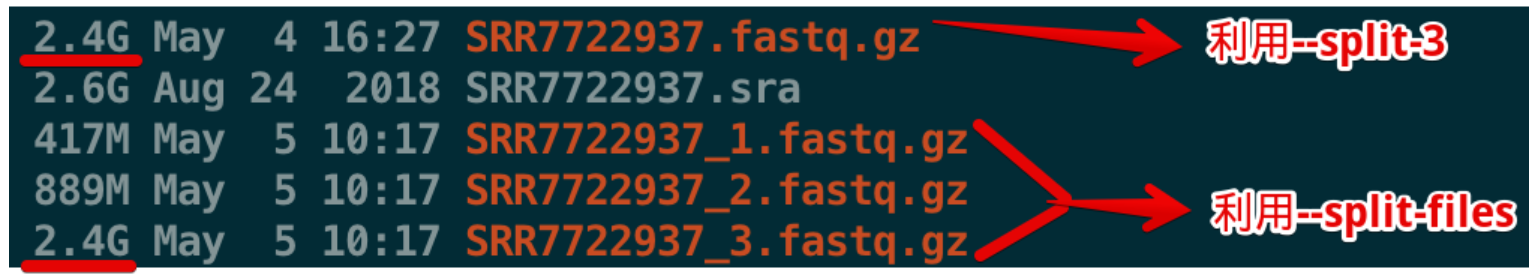
--gzip将生成的结果fastq文件进行压缩--split-3其实有点复杂:首先它是分割的意思,-3实际上指的是分成3个文件,它诞生的时间比较早,是在1000 Genome的Phase1阶段产生的。https://www.biostars.org/p/186741/如果结果发现只有一个文件,说明数据不是双端(第三个文件太大会覆盖前两个);
如果结果有两个文件,说明是双端文件并且数据质量比较高(没有低质量的reads或者长度小于20bp的reads);
如果结果有三个文件,说明是双端文件,但是有的数据质量不高,存在trim的结果,第三个文件的名字一般是:
<srr_id>.fastq, 而且文件也不大,基本可以忽略- -A指定输出的文件名
如果使用上面的参数--split-3,结果只有一个fq文件
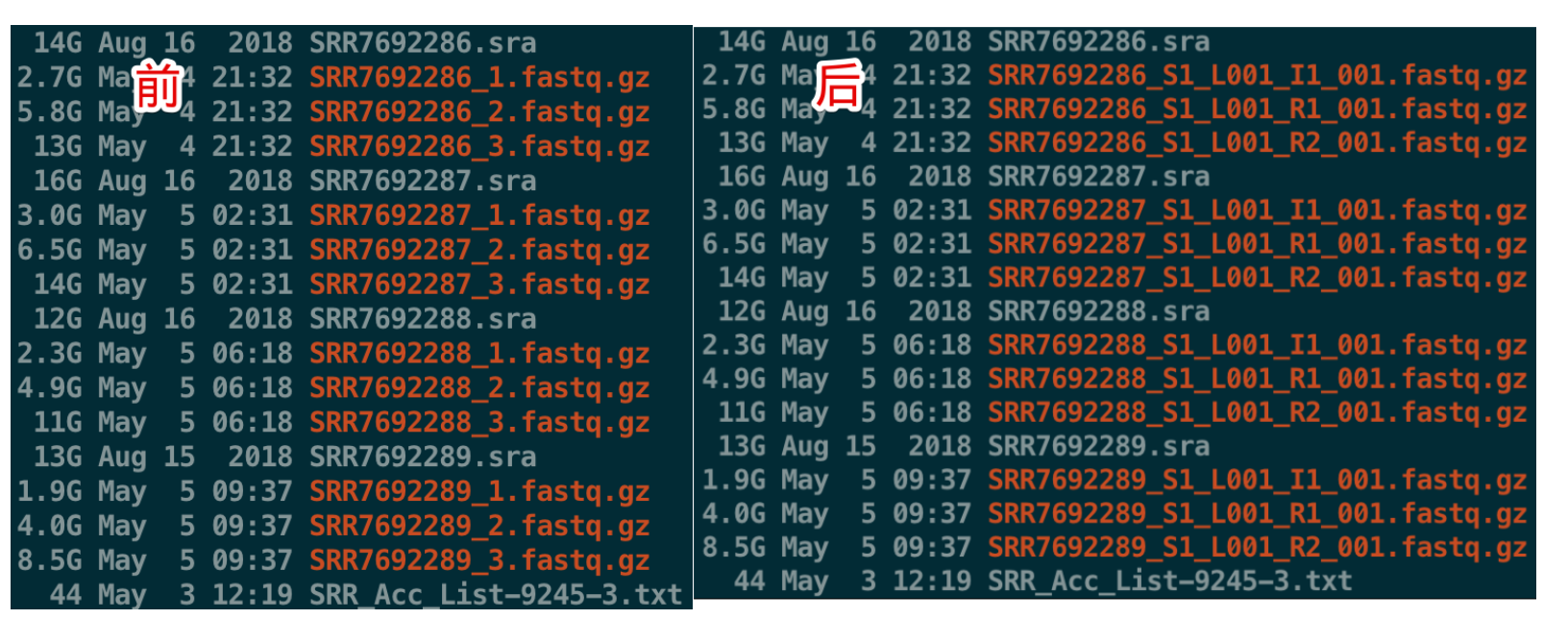
利用cell ranger软件分析,一般需要两个输入文件,其中一个是测序reads,另一个是UMI+Barcode文件,那么只生成一个文件是不够的,因此可以换个参数
使用另外一个参数--split-files来替代--split-3 ,就可以生成三个文件,其中第一个文件的所有序列都是8bp,第二个文件都是26bp,第三个文件都是91bp,初步判断,第三个文件是测序reads
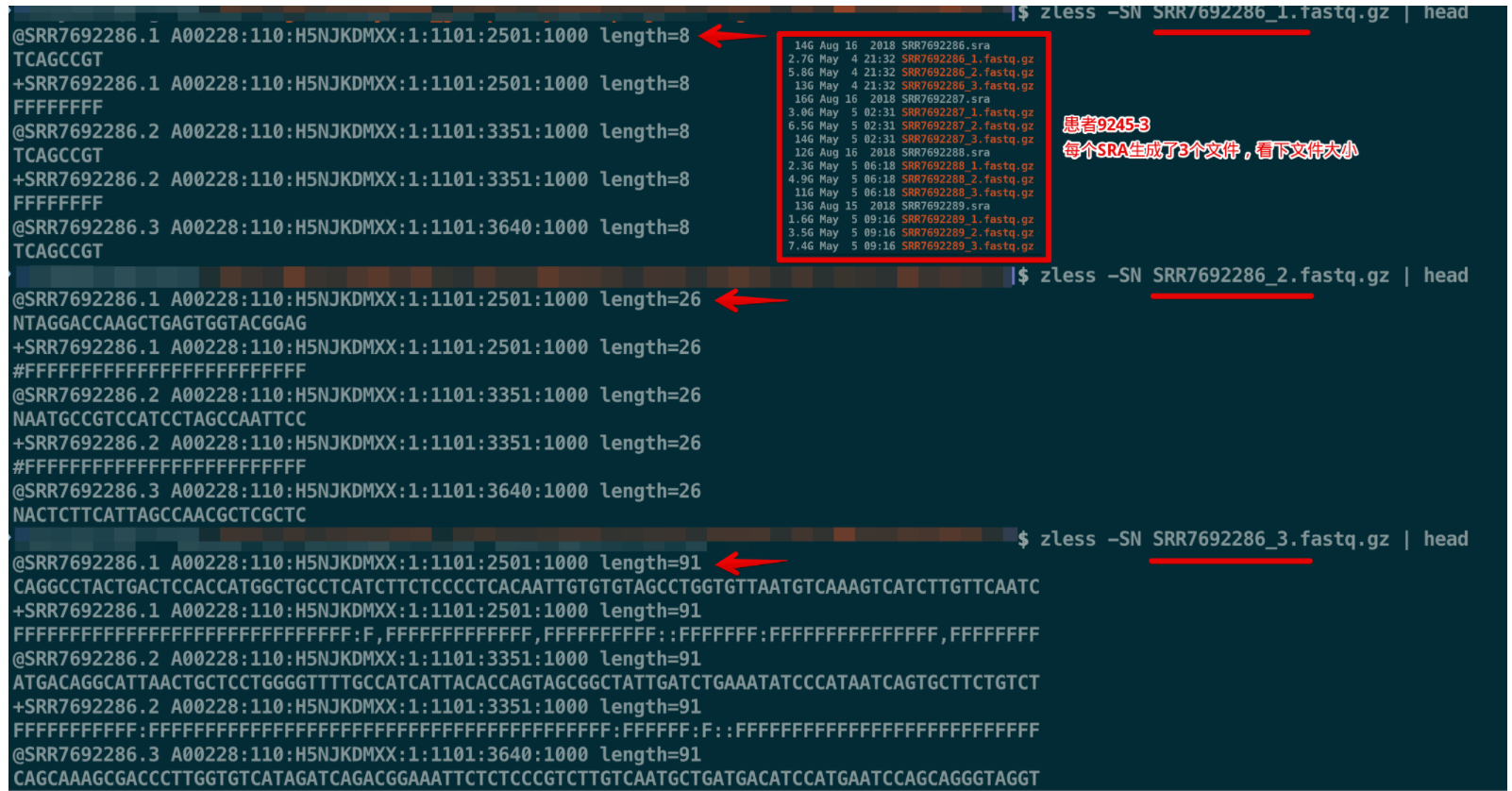
这里可以对比下这两个参数输出的数据
但是前两个呢?哪一个是UMI+Barcode?
如何解释生成的这三个fastq文件
单细胞转录组数据和普通的bulk转录组还是不太一样,bulk结果一般就是R1、R2,很容易区分;10X单细胞数据比较特殊,它的测序文库中包括index、barcode、UMI和测序reads。
文章使用的是10X Genomics 3' Chromium v2.0 平台,那么就看一下它的帮助手册(https://assets.ctfassets.net/an68im79xiti/1CnKSfa7taoQwIEe0WaA4m/8635b2c9ee86c022e731b6fb2e13fed2/CG000080_10x_Technical_Note_Base_Composition_SC3_v2_RevB.pdf )
先大概了解一下10X文库组成:

其中Read2:98那里的星号表示这个长度不是固定的,可以调整,比如文章中患者P2586-4的Read2长度就是98,而患者9245-3的Read2长度是91
然后看看测序时每个run cycle做了什么事:
利用illumina边合成变测序(sequencing by synthesis ,SBS),每一个cycle都是一个碱基,因此用cycle数可以表示测序长度

首先,1-26个cycle就是测序得到了26个碱基,先是16个Barcode碱基,然后是10个UMI碱基;
然后,27-34这8个cycle得到了8个碱基,就是i7的sample index;
最后35-132个cycle得到了98个碱基,就是转录本reads
看下Read1、i7 index、Read2的碱基分布:
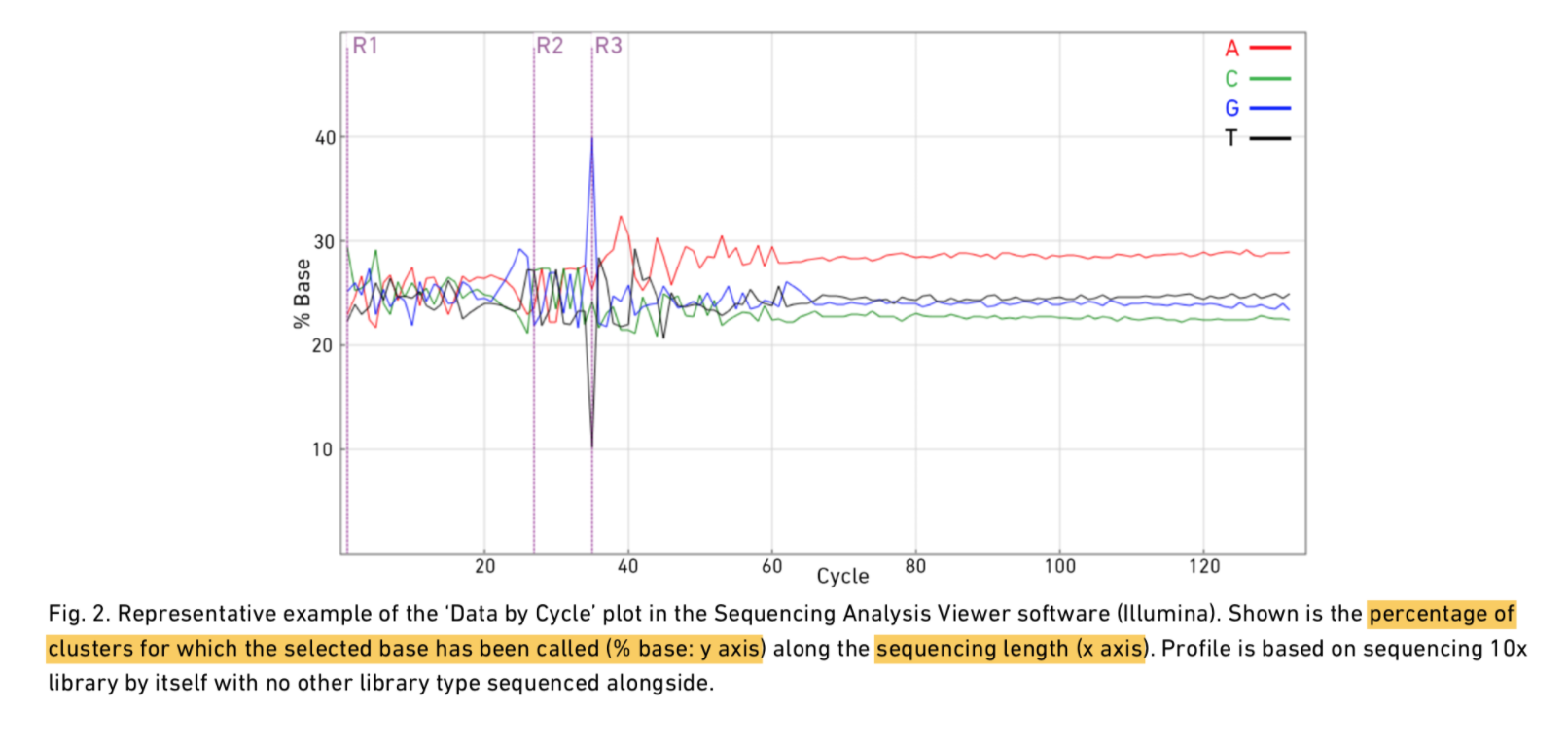
可以看到转录本read前端有20多bp质量是存在波动的,因为5’端的前几个碱基为随机引物序列,存在一定的偏好性
另外,index和barcode有什么区别,为什么用两个fq文件进行区分?
找到10X官方给出的一个解答:https://kb.10xgenomics.com/hc/en-us/articles/115002777072-How-do-I-demultiplex-by-sample-index-and-barcode-
- i7 sample index(library barcode)是加到Illumina测序接头上的,保证多个测序文库可以在同一个flow-cell上或者同一个lane上进行混合测序(multiplexed)。当然可以自己指定index,但更多情况下会使用10X公司提供的index序列(bundled index sets),针对不同项目使用的index也是不同的。不过共性就是:96孔板的每个孔中都加入了4种不同的index oligos混合(详见:https://kb.10xgenomics.com/hc/en-us/articles/218168503-What-oligos-are-in-my-sample-index-)。
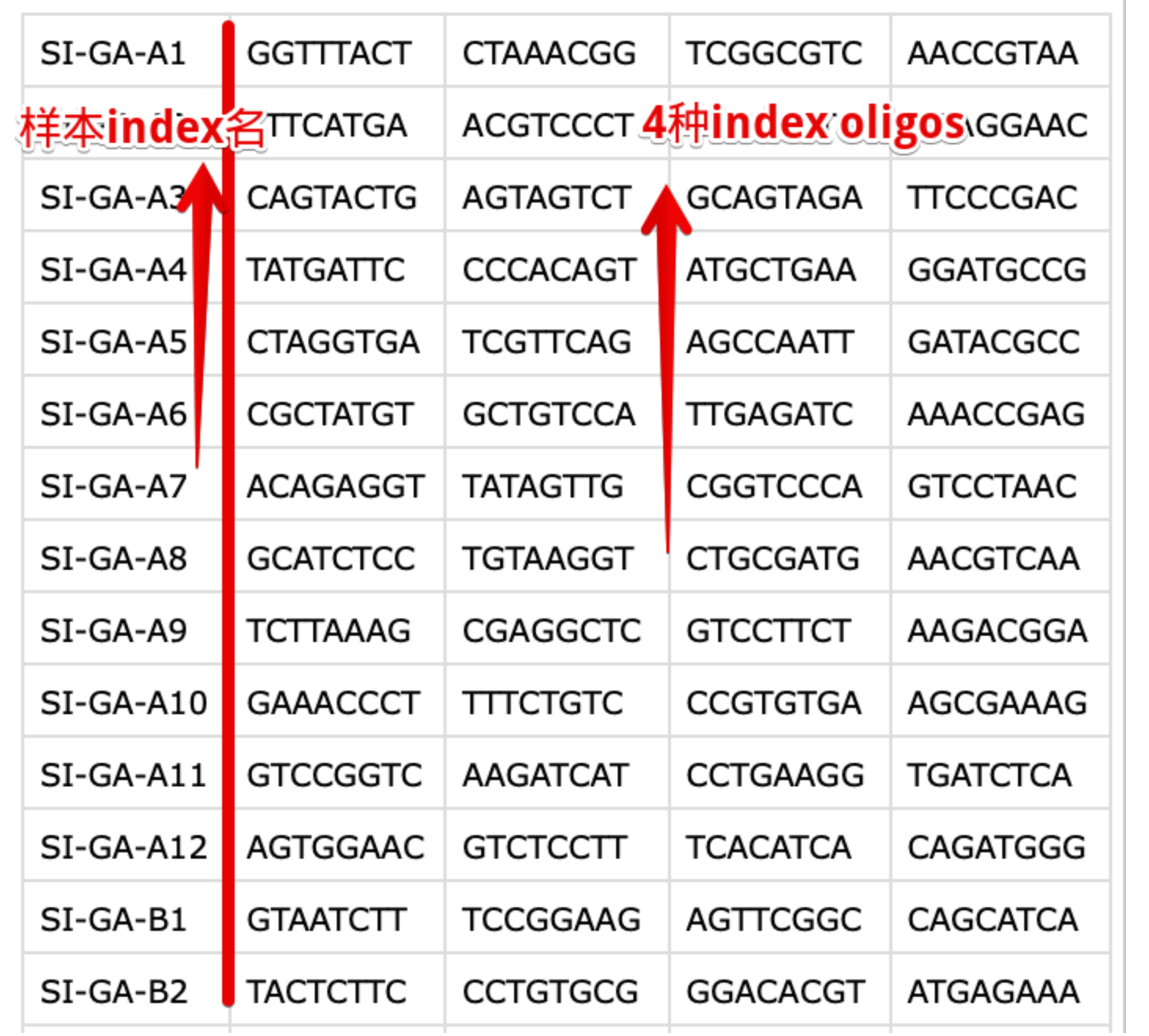
它的作用就是在CellRanger的
mkfastq功能中体现出来的,它自动识别样本index名称(例如:SA-GA-A1),将具有相同4种oligo的fq文件组合在一起,表示同一个样本。它保证了一个测序lane上可以容纳多个样本然后回过头,又看了看GEO的数据记录,发现的确记录了index信息,
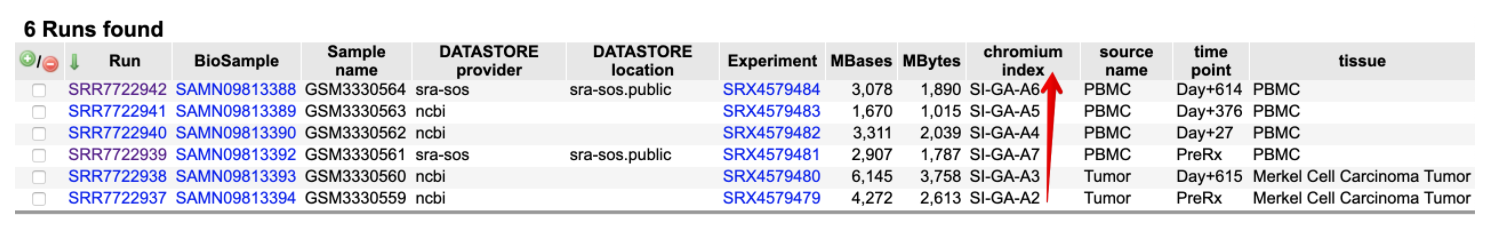
但是作者没有上传最原始的BCL文件,因此无法体验mkfastq的功能,但是官网有测试数据。这里了解大体的拆分流程就好

- 10X Barcode(Cell barcode) 是10X特有的,用来区分GEMs,也就是对细胞做了一个标记。一般在拆分混养测序数据(demultiplexing)这个过程后进行操作,当然这也很符合原文的操作

- 在实验建库的过程中,barcoding过程(也就是GEM生成的过程)是早于indexing过程(它是PCR的最后阶段),但是生信分析过程把这两个顺序颠倒过来了,我们需要先根据样本index,利用
mkfastq将reads进行demultiplex,分到各自的测序文库中去,然后对每个文库再处理barcodes信息
UMI的作用呢?
它是为处理PCR 扩增偏差而生
首先,不管是bulk RNA还是scRNA,都需要进行PCR扩增,但是不可避免有一些转录本会被扩增太多次,超过了真实表达量。当起始文库大小很小时(比如单细胞数据),就需要更多次的PCR过程,这个次数越多,引入的误差就越大
UMI就是Unique Molecular Identifier,由4-10个随机核苷酸组成,在mRNA反转录后,进入到文库中,每一个mRNA随机连上一个UMI,根据PCR结果可以计数不同的UMI,最终统计mRNA的数量。

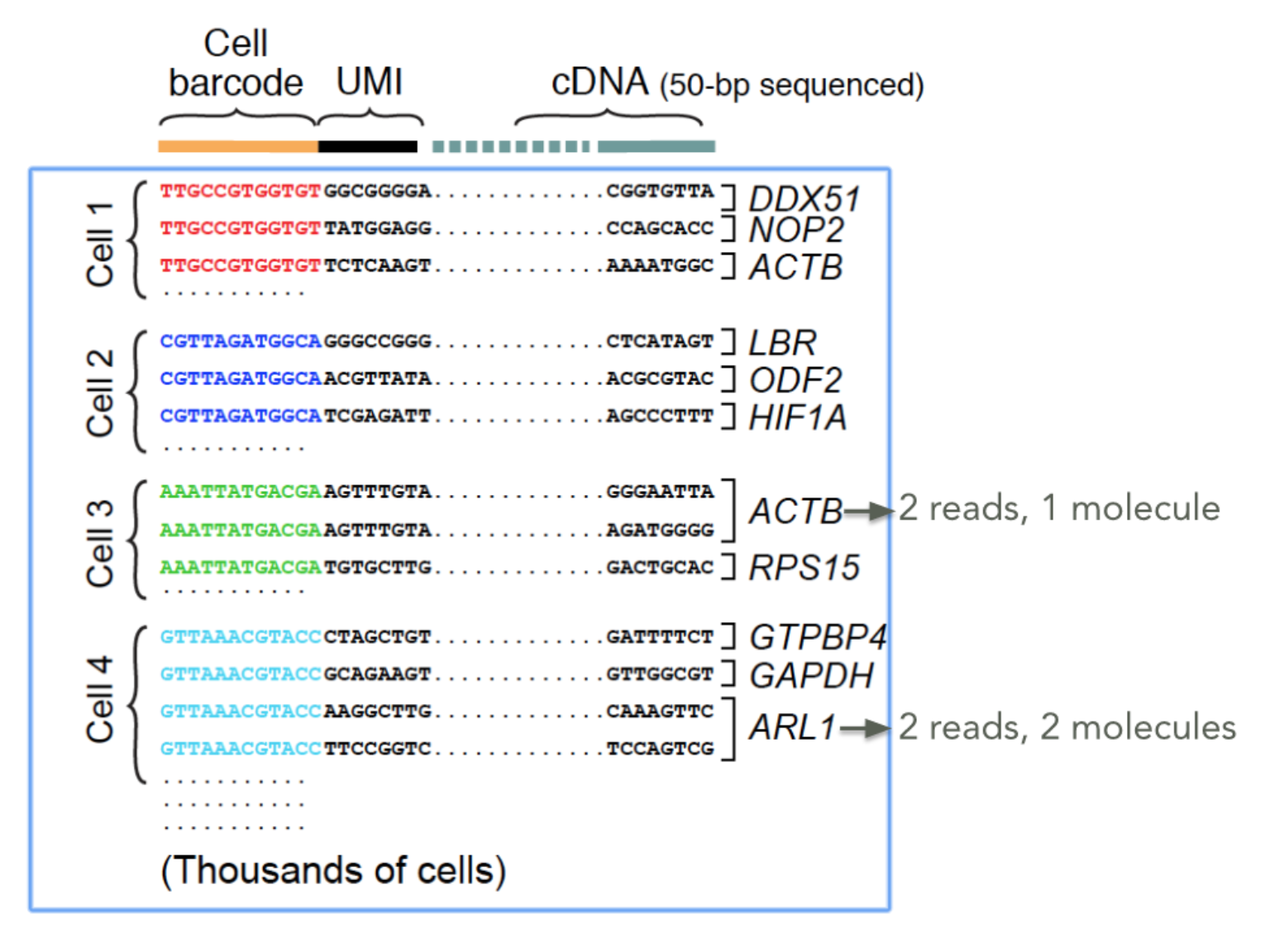
UMI有几个要求:
- 不能是均聚物 ,如AAAAAAAAAA
- 不能有N碱基
- 不能包含碱基质量低于10的碱基
综上所述:raw fastq中包含以下信息
- Library Barcode (Sample Index) - Used to pool multiple samples on one sequencing lane
- Cell Barcode (10x Barcode) – Used to identify the cell the read came from
- Unique Molecular Index (UMI) – Used to identify reads that arise during PCR replication
- Sequencing Reads – Used to identify the gene a read came from
那么这三个文件的名称需要修改吗?
我认为是需要修改的,因为命名太模糊,不容易指定文件进行下游分析。
然后看到官网给出的解答:https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/2.0/using/fastq-input#wrongname ,也的确说需要修改
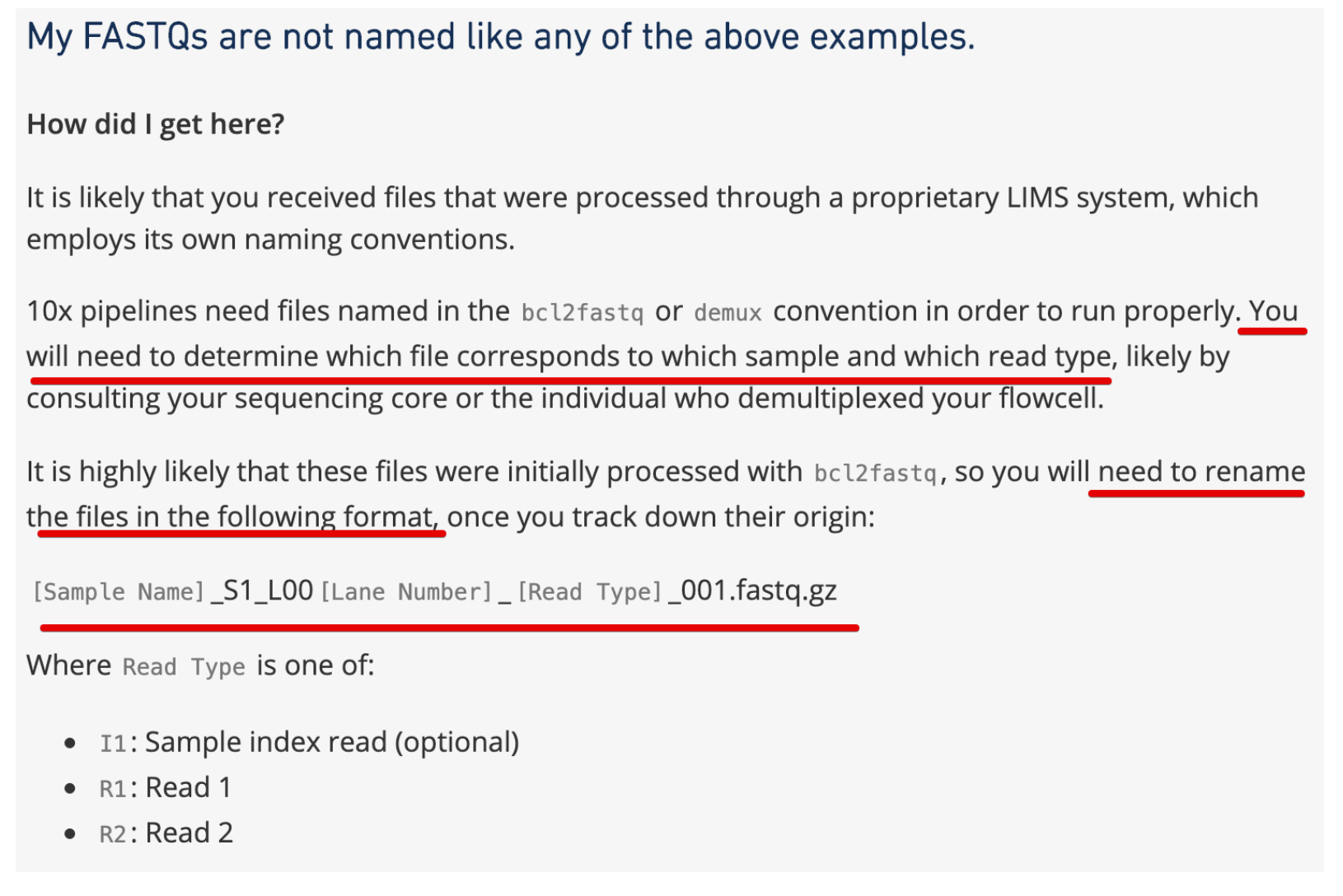
那么怎么改?
肯定要批量处理,就利用下载SRA的SRR ID好了
- # 比如,将原来的SRR7692286_1.fastq.gz改成SRR7692286_S1_L001_I1_001.fastq.gz
- # 依次类推,将原来_2的改成R1,将_3改成R2
- cat SRR_Acc_List-9245-3.txt | while read i ;do (mv ${i}_1*.gz ${i}_S1_L001_I1_001.fastq.gz;mv ${i}_2*.gz ${i}_S1_L001_R1_001.fastq.gz;mv ${i}_3*.gz ${i}_S1_L001_R2_001.fastq.gz);done