

首先对上次改好名称的fastq数据进行质控
- # 以P2586-4为例
- mkdir -p $wkd/qc
- cd $wkd/qc
- find $wkd/raw/P2586-4 -name '*R1*.gz'>P2586-4-id-1.txt
- find $wkd/raw/P2586-4 -name '*R2*.gz'>P2586-4-id-2.txt
- cat P2586-4-id-1.txt P2586-4-id-2.txt >P2586-4-id-all.txt
- cat P2586-4-id-all.txt| xargs fastqc -t 20 -o ./
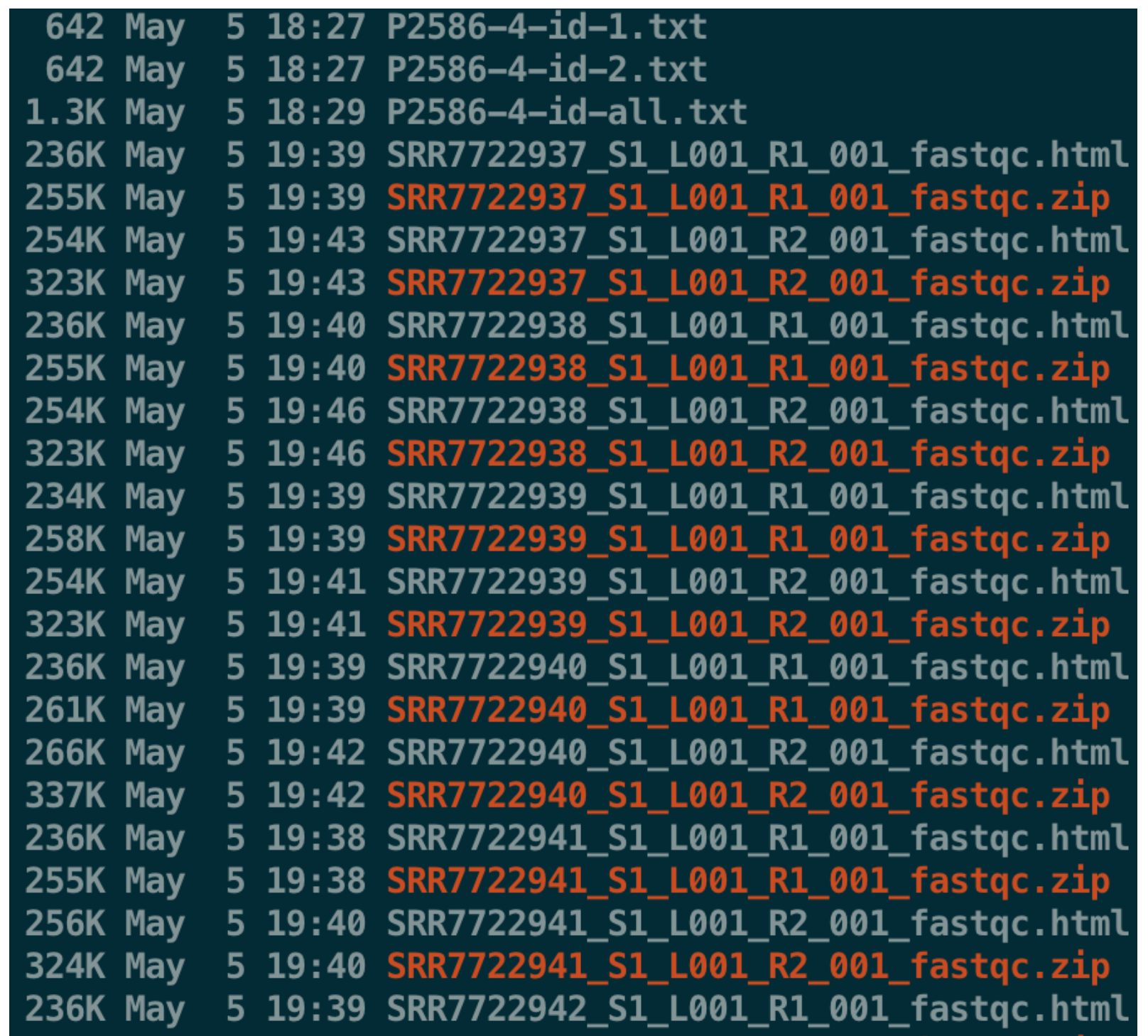
然后利用Filezilla下载其中SRR7722937的R1、R2的html,打开看下
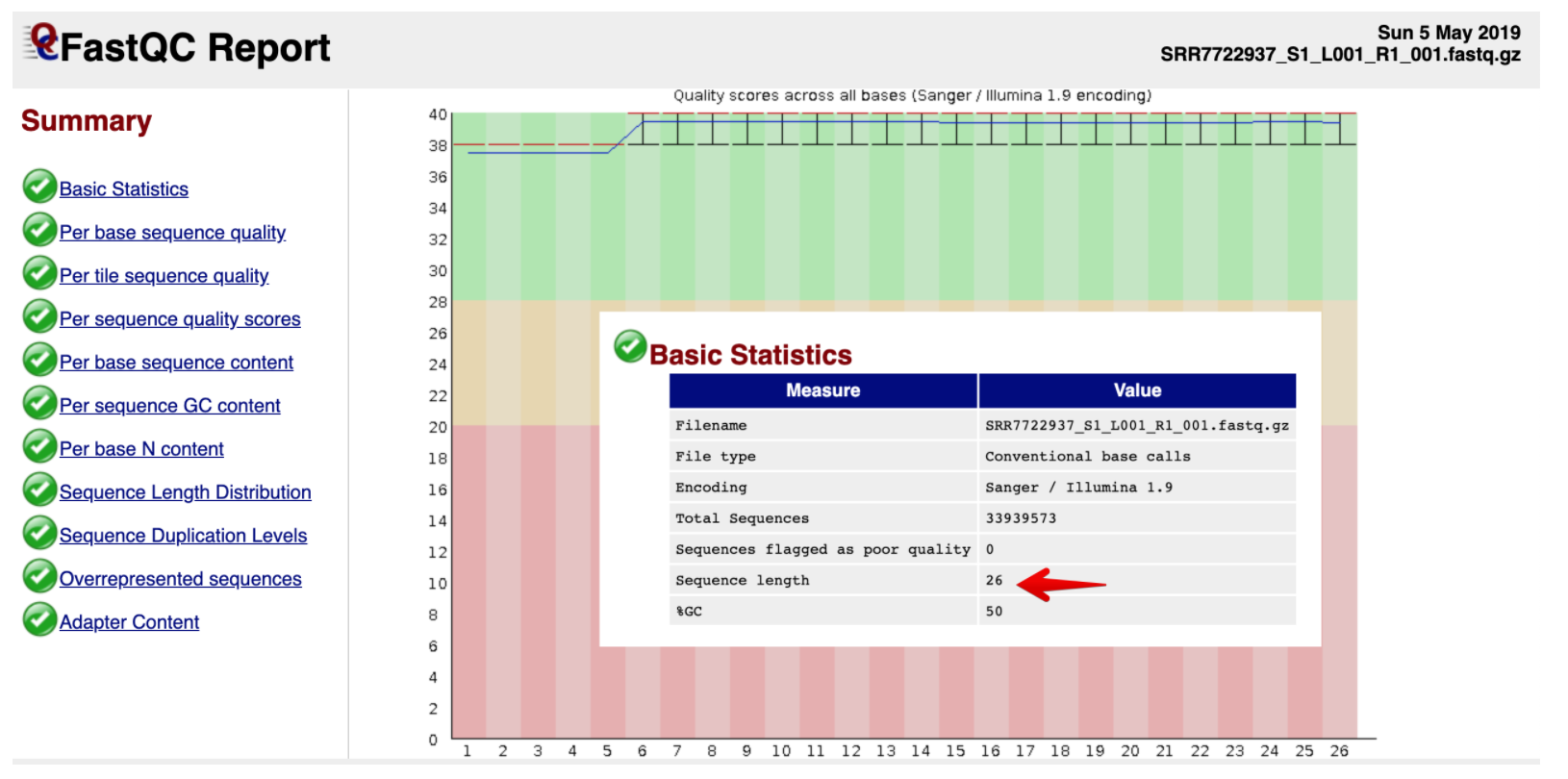
首先是R1的,这个就是16bp Barcode+10bp UMI,可以看到Phred值是比较稳定的
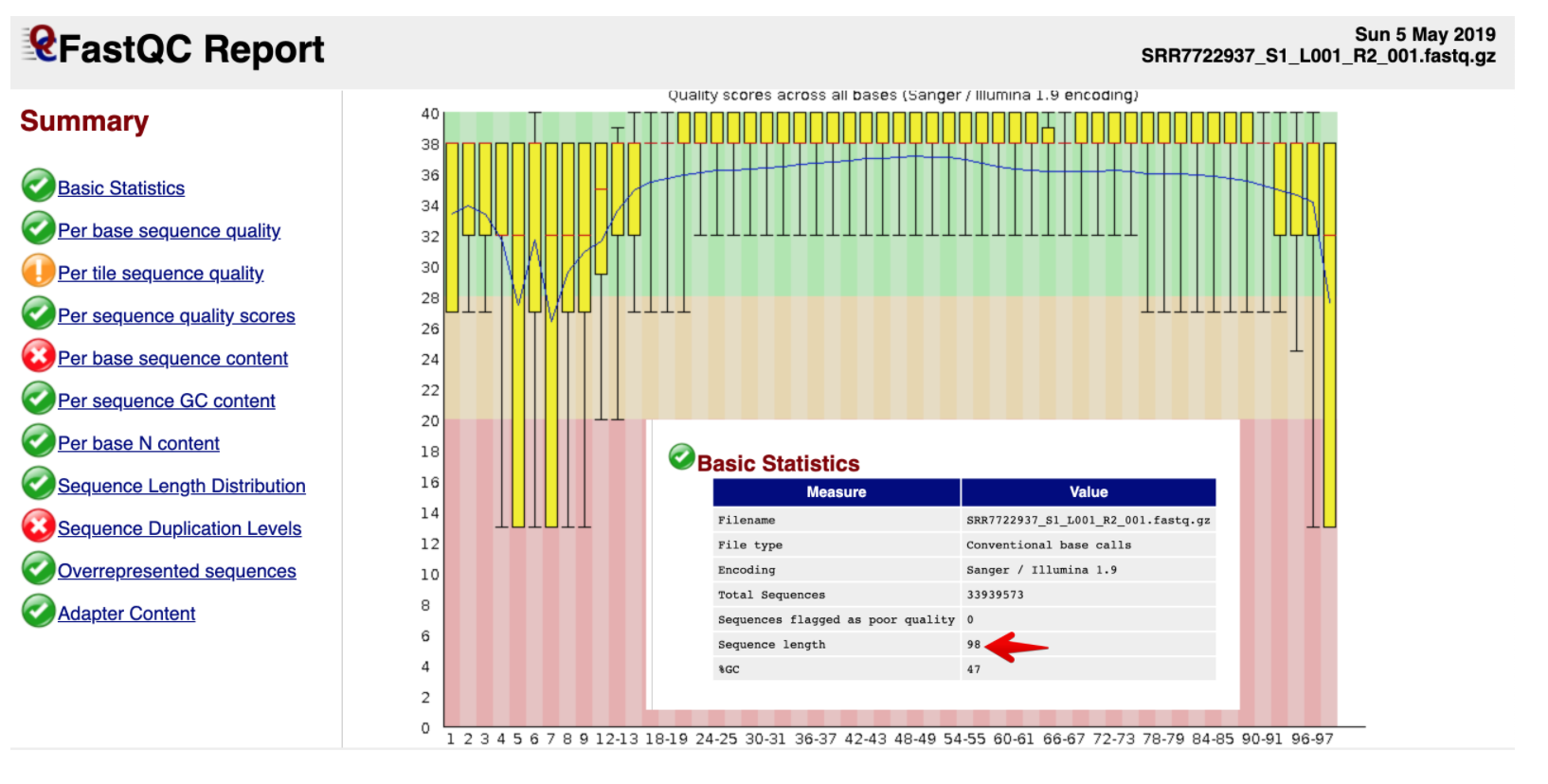
然后看下真实的测序数据:它的Phred值开始质量较差,中间较好,测序结束位置质量有下降,这是和测序仪的工作原理有关的
测序仪在刚开始进行合成反应的时候也会由于反应还不够稳定,会带来质量值的波动;碱基的合成利用的的是聚合酶化学反应,使得碱基可以从5’端向3’端合成并延伸。但合成的过程中随着链的增长,DNA聚合酶的效率会不断下降,特异性也开始变差,越到后面碱基合成的错误率就会越高)
总体上还是在Q30以上的,数据质量不错,并且没有接头序列
如果要对Fastqc结果进行详细的了解,可以去官网的帮助信息,非常的简明扼要:<https://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/3%20Analysis%20Modules/
Cell Ranger的介绍
什么是Cell Ranger?
官网的说明最原汁原味:
Cell Ranger is a set of analysis pipelines that process Chromium single cell 3’ RNA-seq output to align reads, generate gene-cell matrices and perform clustering and gene expression analysis.
到目前为止,它有1.0,1.1,1.2,1.3,2.0,2.1,2.2,3.0这8个版本,其中1.2版本之后的可以支持Chromium Single Cell 3' v1 and v2 试剂,最新的V3试剂需要用到3.0版本
要分析的文章中使用了两个不同的版本(2.0和2.1)去分析两个患者的数据:

那么这个软件能干什么事?
它主要包括四个主要基因表达分析流程:
- cellranger mkfastq : 它借鉴了Illumina的
bcl2fastq,可以将一个或多个lane中的混样测序样本按照index标签生成样本对应的fastq文件 - cellranger count :利用
mkfastq生成的fq文件,进行比对(基于STAR)、过滤、UMI计数。利用细胞的barcode生成gene-barcode矩阵,然后进行样本分群、基因表达分析 - cellranger aggr :接受
cellranger count的输出数据,将同一组的不同测序样本的表达矩阵整合在一起,比如tumor组原来有4个样本,PBMC组有两个样本,现在可以使用aggr生成最后的tumor和PBMC两个矩阵,并且进行标准化去掉测序深度的影响 - cellranger reanalyze :接受
cellranger count或cellranger aggr生成的gene-barcode矩阵,使用不同的参数进行降维、聚类
它的结果主要是包含有细胞信息的BAM, MEX, CSV, HDF5 and HTML文件
相关的术语
- Sample:(样本)从单一来源(比如血液、组织等)提取的细胞悬液
- Library:(文库)单个样本制备的10X barcode 测序文库,对应10X Chromium Controller一个run(即运行一次)的单个芯片通道
- Sequencing Run / Flowcell: 测序仪主要依靠flowcell(例如Hiseq4000就有2个flowcell),每运行一次就是run一次,数据会在flowcell上产出,然后这些数据又会根据flowcell上的不同lane以及lane上不同样本的index进行区分
- 以上术语的错综复杂:
- 单独一个样本可以制备成多个文库,这样可以一次run就得到更多的细胞数量,而不用在单次run的单个文库中使用全部的样品,造成"过载"现象("过犹不及")
- 单独一个样本可以跨多个flowcell测序,如果它们是在一次测序过程中产出,就可以将产出的reads合并
- 上面说一个文库可以用多个flowcell,那么一个flowcell也可以包含多个文库,使用不同的lane上或者样本的index进行区分
- 同一个10x 芯片的两个channels可以说是两个文库,但是两个不同芯片的同一个channel不能说是两个文库
大体流程
主要根据sample、library、flowcell的数量来定义分析的复杂程度(由浅入深)
- 一个sample、一个library、一个flowcell
这是最基本的模式:只有一个生物样本,只需要制备一个文库,在一个flowcell上测序就好,通过
mkfastq得到fastq文件后直接运行count就好(具体见: Single-Library Analysis) - 一个library,多个flowcell
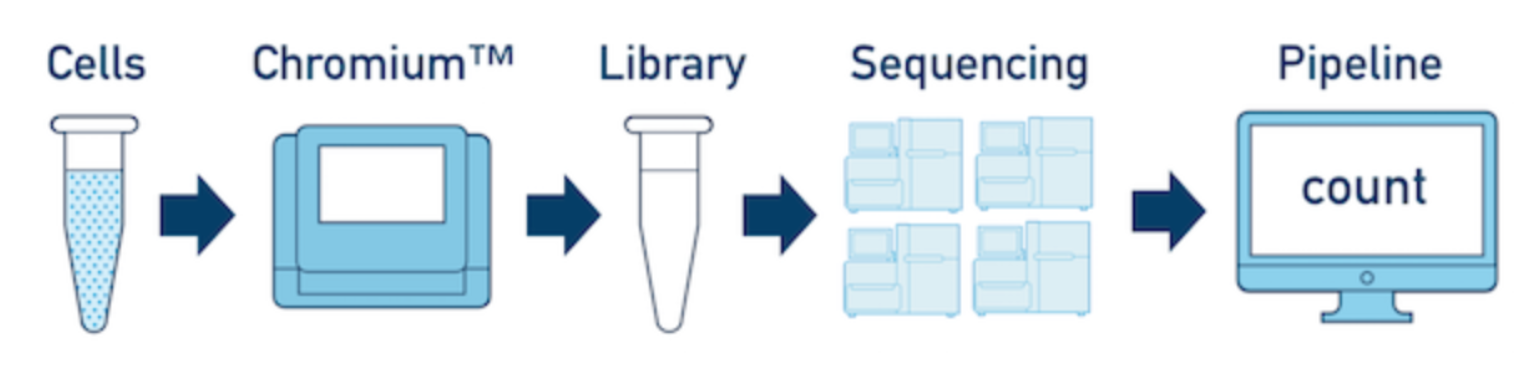
如果有个文库在多个flowcell上测序,可以利用 Running Multi-Library Samples 将不同测序run的reads混合
- 一个sample,多个library
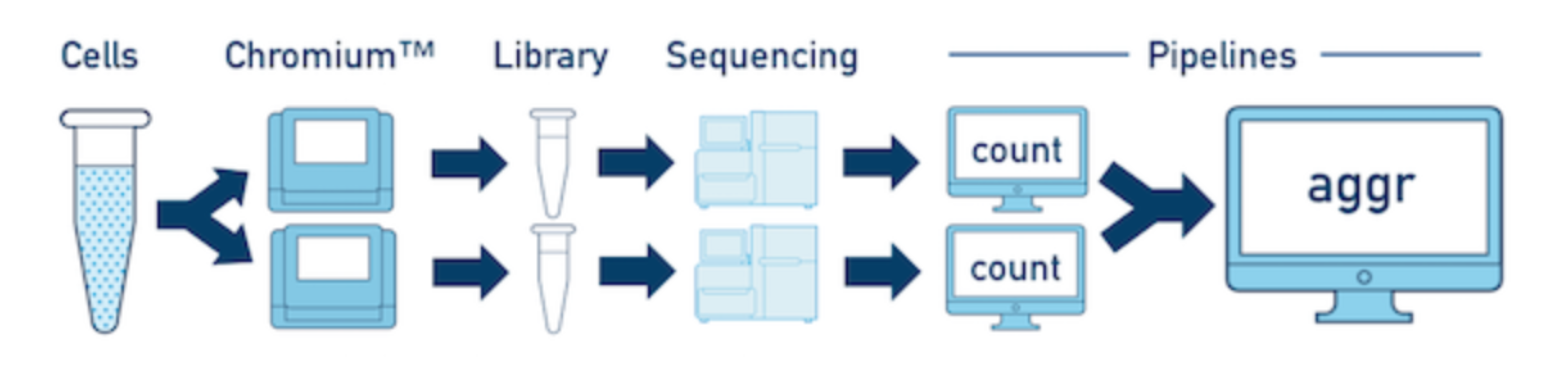
如果一个样本有多个文库(比如做了技术重复),那么每一个文库的数据应该单独跑
cellranger count流程,然后可以利用aggr整合起来, Multi-Library Aggregation ;
当然先将一个样本的多个文库组合在一起再分析也是可以的,就需要利用到 MRO syntax (这里先做了解) - 多个samples
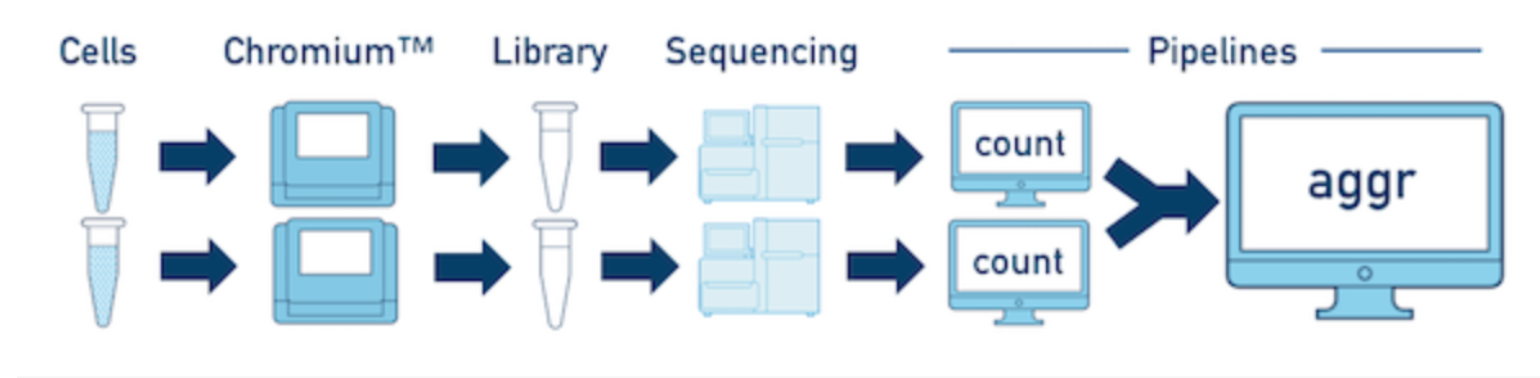
必须每个样本的每个文库单独跑
cellranger count,比如说:实验中有4个样本,每个样本由两个文库(或者两个技术重复),那么就需要跑count流程8次,最后利用aggr汇总在一起
Cell Ranger的安装与配置
系统要求
- Linux 至少8核CPU(推荐16核)
- 64GB RAM(推荐128GB),最低配的64G允许
cellranger aggr合并最多250k个细胞 - 1T硬盘
- 64-bit CentOS/RedHat 6.0 or Ubuntu 12.04
软件依赖
大多软件是和Cell Ranger打包在一起的,但是cellranger mkfastq 依然需要Illumina bcl2fastq (要求版本2.17以上;如果使用Novaseq,最好用版本2.20或者更高)
使用资源限制
Cell Ranger默认在本地运行(或者使用--jobmode=local指定),它会占用90%的空余内存以及所有空余的CPU。如果要进行资源限制,可以使用—localmem或者--localcores
使用
localcores之前,应该先检查ulimit -u,看看服务器最大支持多少用户同时在线,因为cellranger使用一个核就会产生64个用户队列,不能超过这个限定值。例如检查ulimit -u为4096,那么最多设置64个核心,也就是64 * 64 = 4096,才不会因为这个报错
下载软件
为了复现文章,我们需要用到Cell Ranger 2.0与2.1,其中2.0版本是2017年9月发布的,2.1版本是2018年2月发布的
- # 2.0版本下载(732M)
- curl -o cellranger-2.0.2.tar.gz "http://cf.10xgenomics.com/releases/cell-exp/cellranger-2.0.2.tar.gz?Expires=1557256518&Policy=eyJTdGF0ZW1lbnQiOlt7IlJlc291cmNlIjoiaHR0cDovL2NmLjEweGdlbm9taWNzLmNvbS9yZWxlYXNlcy9jZWxsLWV4cC9jZWxscmFuZ2VyLTIuMC4yLnRhci5neiIsIkNvbmRpdGlvbiI6eyJEYXRlTGVzc1RoYW4iOnsiQVdTOkVwb2NoVGltZSI6MTU1NzI1NjUxOH19fV19&Signature=HoJUuPo4iTFdQgzFU1GH7uKf3uGitQxTjB6WOA9qGPlejf7tNcBPjO65WuSUZ~w8WWdeAvky-oV7XGfheY-bUr2b7QHr7jQEqc84cyU~PLvT~fYjkgC7cG7nlpbJOT~b7U~YH9amvR~SCLlyynp7scPDIA~9~keCYrIPgevTf2QyktybuSyjNTwugefOic~~XFkc9lrS~WQ9MNA1CLl4ExlQKsxWS77PEB6mwrMZXX65obDnZW9fIs3dIny6H5YoadbkgmsT52jmLien6PsG1g2jpAO90pPuHoru8LL64Q9gmB3I0nJAqi3EmrO3GKnUpHUhGb6doKmjSN6XccpmsA__&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA"
- # 2.1版本下载
- curl -o cellranger-2.1.1.tar.gz "http://cf.10xgenomics.com/releases/cell-exp/cellranger-2.1.1.tar.gz?Expires=1557260110&Policy=eyJTdGF0ZW1lbnQiOlt7IlJlc291cmNlIjoiaHR0cDovL2NmLjEweGdlbm9taWNzLmNvbS9yZWxlYXNlcy9jZWxsLWV4cC9jZWxscmFuZ2VyLTIuMS4xLnRhci5neiIsIkNvbmRpdGlvbiI6eyJEYXRlTGVzc1RoYW4iOnsiQVdTOkVwb2NoVGltZSI6MTU1NzI2MDExMH19fV19&Signature=RNQd-gTASTQhtnUSBfQWrnqo6Pyy2wDXtV5tlxkG97727GvoRhMqFXbEsz4gJl2BMckdVvW3S1tZRwRo5pmxPzmhq-8RKxf99pGqlzo84HYqhbIRkxXlIbLbj-u3PUJqo8cesWpbSVSKkS2TCNS-9GMFNieQswqMS2-DN4BqoBOJnWr7T4wlOMd9hypXWwOsW2P2fqaM-WP2ooMyo-oIxm3y9gDghXdDEP5lvHU7GCQcFGGexkdIrD6S5p8JPJ1DB5XieGrtEuP1YVp6tLMGXFoRWXS8dQLI1egWDYlOuRaiQgLIb3o5ZxBg5NpzLPP5kDHMAVzJFdBpf~~rkyNYTA__&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA"
软件下载完,需要解压缩一段时间,因为它打包了太多的软件和资源
- tar zxvf cellranger-2.0.2.tar.gz
然后添加环境变量(注意:如果之前安装过其他版本,比如我之前装过3.0,但是当前你只想用这个2.0版本,那么就需要在~/.bashrc中将新安装的2.0版本路径放在3.0的下方,因为linux是根据$PATH自上而下调用软件的,将新安装的路径放在.bashrc下方,那么在$PATH中显示的就是新路径在上方,它们的顺序是相反的)
- # 举个例子
- # 原来我的~/.bashrc中有个cell ranger 3.0
- echo 'export PATH=/home/biosoft/cellranger-2.2.0:$PATH' >> ~/.bashrc
- # 现在我想输入cellranger时先调用2.0版本,那就在3.0的下方写上
- export PATH=/home/biosoft/cellranger-3.0:$PATH
- export PATH=/home/biosoft/cellranger-2.0.2:$PATH
- # 然后保存退出,激活环境变量
- # 这时查看环境变量
- echo $PATH
- /home/biosoft/cellranger-2.0.2:/home/biosoft/cellranger-3.0
- # 于是输入cellranger,给出的帮助文档就是
- cellranger (2.0.2)
- Copyright (c) 2017 10x Genomics, Inc. All rights reserved.
- -------------------------------------------------------------------------------
- Usage:
- cellranger mkfastq
- cellranger count
- cellranger aggr
- cellranger reanalyze
- cellranger mkloupe
- cellranger mat2csv
- cellranger mkgtf
- cellranger mkref
- cellranger vdj
- cellranger mkvdjref
- cellranger testrun
- cellranger upload
- cellranger sitecheck
- # 成功切换了版本!
然后安装好以后,cellranger还提供了一个小工具(我认为是个有意思的地方),让你全面了解你的linux性能,不用自己找代码。另外这些代码我们也可以借鉴,以后再用
- $ cellranger sitecheck > sitecheck.txt
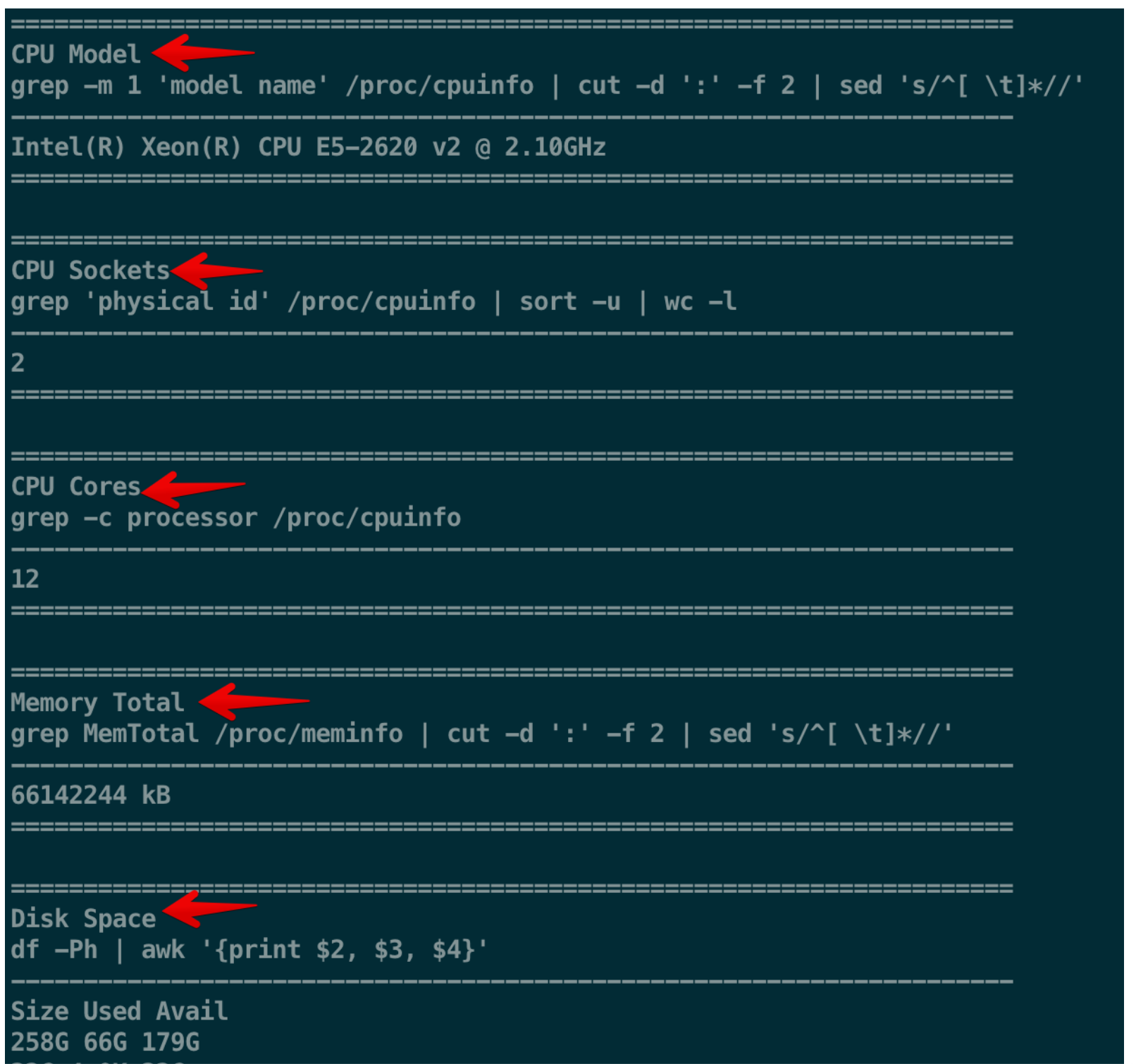
为了确保软件所有的自带流程都成功安装,可以进行一个软件自检
- cellranger testrun --id=tiny
- # 我使用了12个CPU,大约需要20分钟检查完
- # 如果成功完整地安装的话,最后会给出这样一个报告:
- cellranger testrun (2.0.2)
- Copyright (c) 2017 10x Genomics, Inc. All rights reserved.
- -------------------------------------------------------------------------------
- Running Cell Ranger in test mode...
- Martian Runtime - 2.0.2-2.2.2
- Running preflight checks (please wait)...
- [runtime] (ready) ID.tiny.SC_RNA_COUNTER_CS.SC_RNA_COUNTER.SETUP_CHUNKS
- [runtime] (split_complete) ID.tiny.SC_RNA_COUNTER_CS.SC_RNA_COUNTER.SETUP_CHUNKS
- ...
- Pipestance completed successfully!
参考序列下载(1.2.0版本,2016年12月发布)
文章比对到了hg38(基于ensembl数据库),不能直接使用网站下载的基因组与注释文件,需要过滤一下
如果直接下载的话一共11GB,当然这里面包含了基因组、注释源文件,以及cell ranger自己利用mkgtf构建的注释和mkref构建的基因组
- curl -O http://cf.10xgenomics.com/supp/cell-exp/refdata-cellranger-GRCh38-1.2.0.tar.gz
- # 然后解压
- tar -xzvf refdata-cellranger-GRCh38-1.2.0.tar.gz
如果要自己尝试构建,可以使用
- # 下载基因组
- wget ftp://ftp.ensembl.org/pub/release-84/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
- gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
- # 下载注释
- wget ftp://ftp.ensembl.org/pub/release-84/gtf/homo_sapiens/Homo_sapiens.GRCh38.84.gtf.gz
- gunzip Homo_sapiens.GRCh38.84.gtf.gz
- # 软件构建注释(可以自选attribute)
- # mkgtf <input_gtf> <output_gtf> [--attribute=KEY:VALUE...]
- cellranger mkgtf Homo_sapiens.GRCh38.84.gtf Homo_sapiens.GRCh38.84.filtered.gtf
- --attribute=gene_biotype:protein_coding
- --attribute=gene_biotype:lincRNA
- --attribute=gene_biotype:antisense
- --attribute=gene_biotype:IG_LV_gene
- --attribute=gene_biotype:IG_V_gene
- --attribute=gene_biotype:IG_V_pseudogene
- --attribute=gene_biotype:IG_D_gene
- --attribute=gene_biotype:IG_J_gene
- --attribute=gene_biotype:IG_J_pseudogene
- --attribute=gene_biotype:IG_C_gene
- --attribute=gene_biotype:IG_C_pseudogene
- --attribute=gene_biotype:TR_V_gene
- --attribute=gene_biotype:TR_V_pseudogene
- --attribute=gene_biotype:TR_D_gene
- --attribute=gene_biotype:TR_J_gene
- --attribute=gene_biotype:TR_J_pseudogene
- --attribute=gene_biotype:TR_C_gene
- # 我看到这里写了这么多gene_biotype(也就是基因的生物类型)的键值对,不禁好奇,GTF中存在多少种基因类型以及对应的数目?
- $ cat Homo_sapiens.GRCh38.84.filtered.gtf |grep -v "#" |awk -v FS='gene_biotype ' 'NF>1{print $2}'|awk -F ";" '{print $1}'|sort | uniq -c
- 213 "IG_C_gene"
- 33 "IG_C_pseudogene"
- 152 "IG_D_gene"
- 76 "IG_J_gene"
- 9 "IG_J_pseudogene"
- 1209 "IG_V_gene"
- 646 "IG_V_pseudogene"
- 125 "TR_C_gene"
- 16 "TR_D_gene"
- 316 "TR_J_gene"
- 12 "TR_J_pseudogene"
- 848 "TR_V_gene"
- 110 "TR_V_pseudogene"
- 45662 "antisense"
- 58181 "lincRNA"
- 2337766 "protein_coding"
- # 软件利用构建好的注释,去构建需要的基因组
- cellranger mkref --genome=GRCh38
- --fasta=Homo_sapiens.GRCh38.dna.primary_assembly.fa
- --genes=Homo_sapiens.GRCh38.84.filtered.gtf
- --ref-version=1.2.0