

Cellranger的结果力求与常规分析的结果格式相同,比如它生成的 barcoded BAM files 也可以放到IGV中查看比对质量
count结果一般放在out目录下,主要有summary和analysis两大类,包含以下几项:
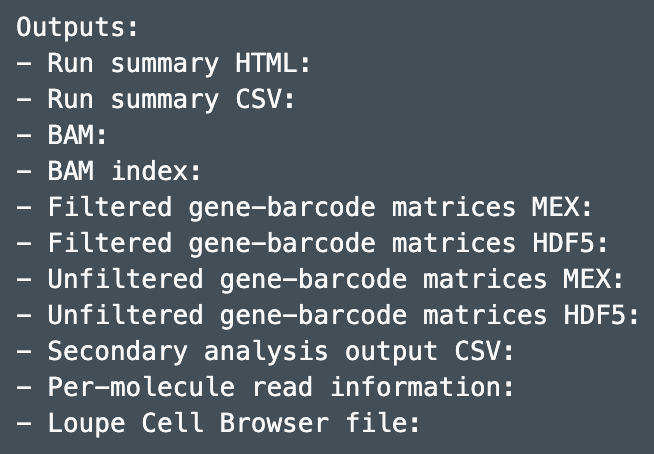
Summary
这是一个html格式的文件,直接下载到本地打开
打开时就能对数据进行一个判断,网页顶端颜色显示为黄色或者红色说明数据存在异常
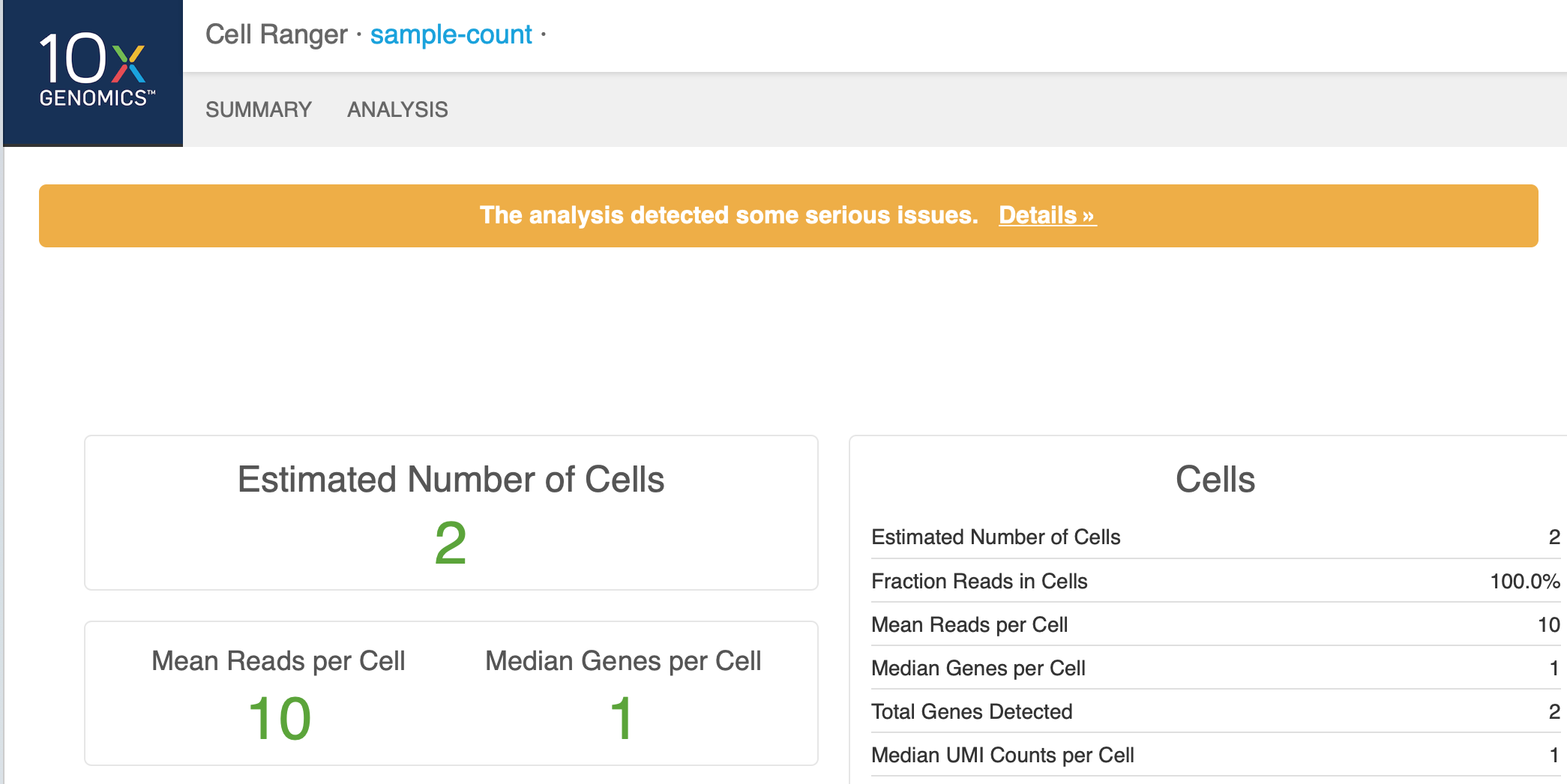
然后点击Details,可以看到为何数据出错:
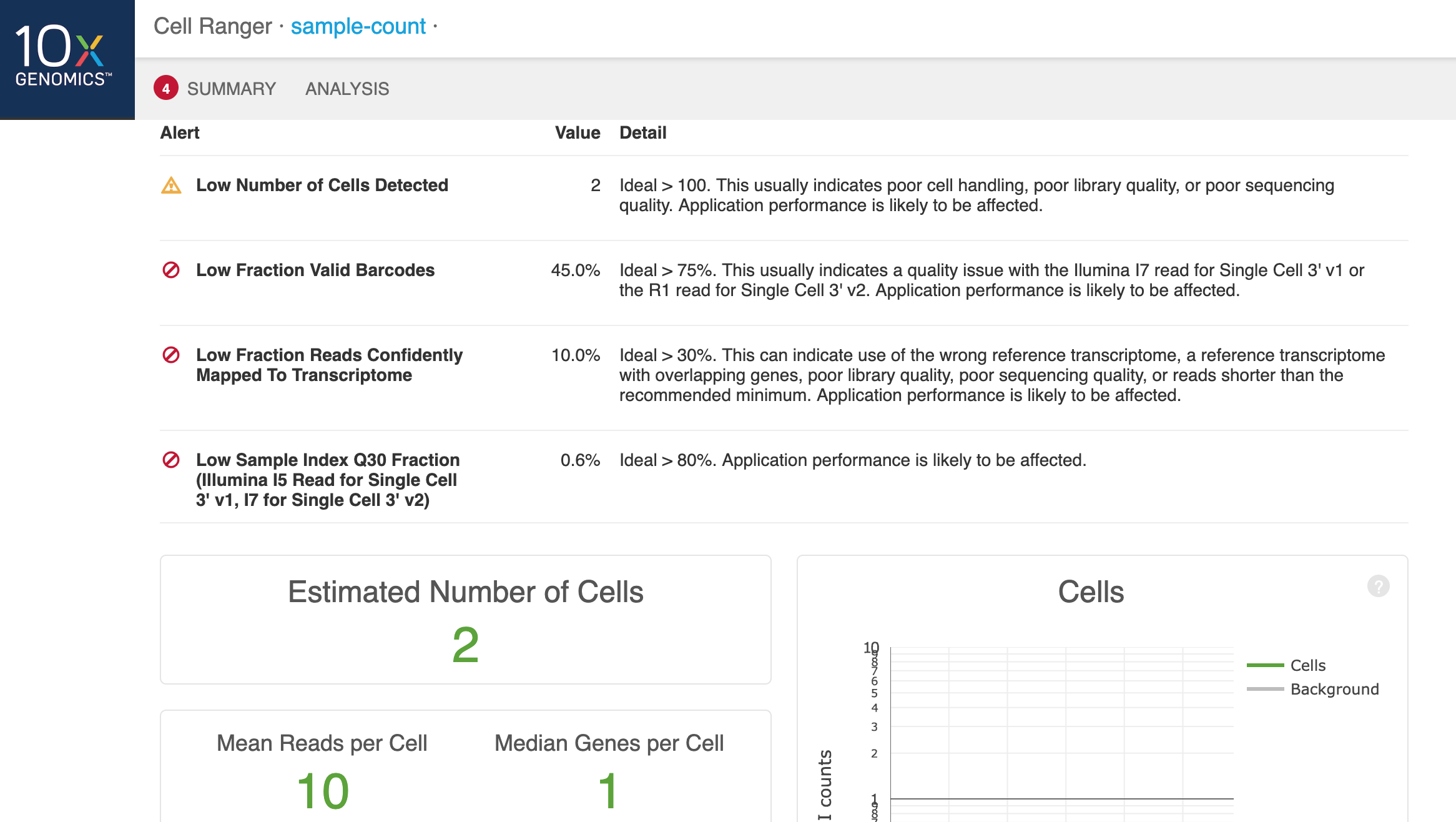
结果部分包括实验捕获的细胞数目、检测到的基因数目、测序数据的产出与质量统计、参考基因组的比对情况
(第一张为V2试剂的结果)
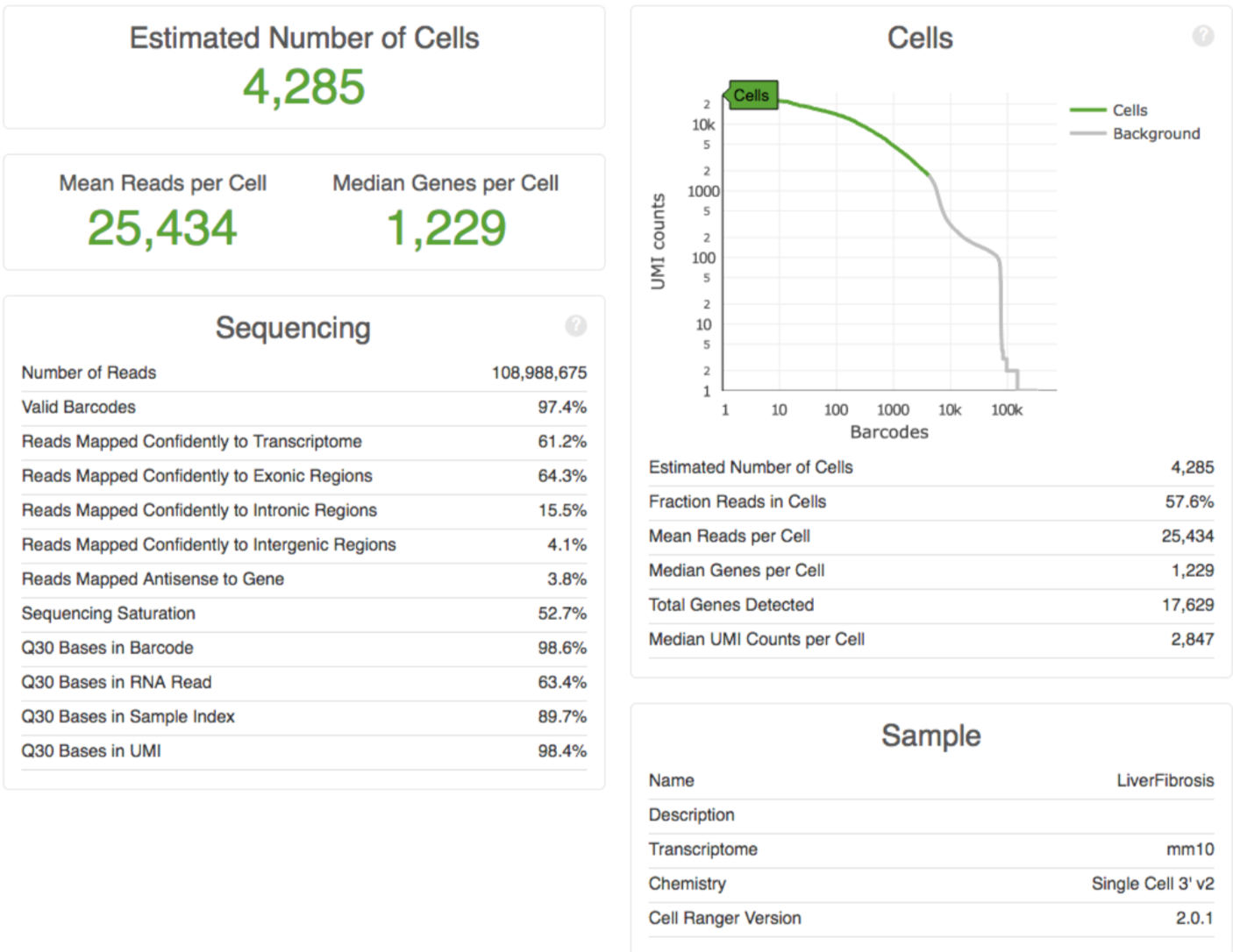
(第二张为V3试剂的结果)
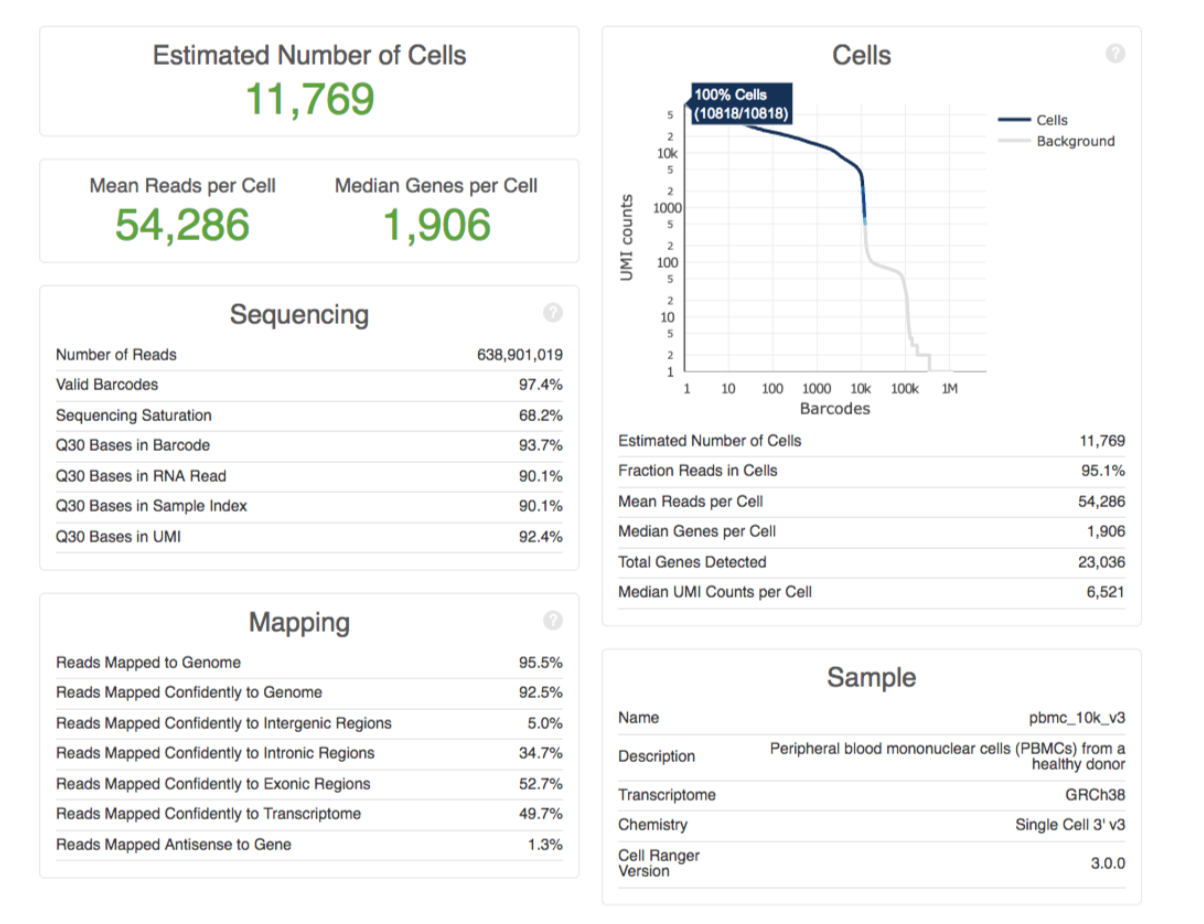
几个指标可以关注一下:
- 左上部分中,包括了reads数、barcodes数、UMI、index、Q30等统计值
- 左下是reads比对的比例,包括基因间区、外显子、内含子,如果比对率太低(一般认为外显子的比对率要在60%以上)
- 右上图是利用barcodes上的UMI标签分布来估计细胞数,绿/蓝色表示细胞,灰色表示背景,其中Y轴表示每个barcode对应的UMI数量,X轴是一定数量的UMI序列所对应barcode的数量,比如上图中有1000个barcodes含有10k个UMI,细胞过滤就是通过这个图来展示的。首先明确,barcode用来区分细胞,UMI用来区分转录本;其次,barcodes数量时要大于细胞数量的(以保证每个细胞都会有barcode来进行区分)如果图线陡降说明
下面👇是在对原文数据进行count时遇到的一个问题,记录下来
原文的小坑
其实有了参考信息与原始序列,跑一个count命令并不难,但是对于这篇文章来讲,有一个小坑需要注意:看原文的方法描述

原文是需要将hg38和Merkel cell polyomavirus, MCPyV(默克尔细胞多瘤病毒)的基因组共同作为参考序列去比对,基本操作流程类似这样:

其中,hg38的fasta与gtf获取比较容易,直接下载即可(具体见"单细胞实战—四"),但是作者还用到了这个MCPyV,它的基因组也是比较容易获得https://www.ncbi.nlm.nih.gov/nuccore/NC_010277,但是gtf怎么获得是个问题,作者给出的解答是:需要用基因组fasta自己生成gtf文件

这个思路很重要,但是如何将基因组fasta转为gtf,还是个问题,于是本着"自己的锅自己背"的原则,又去问了作者
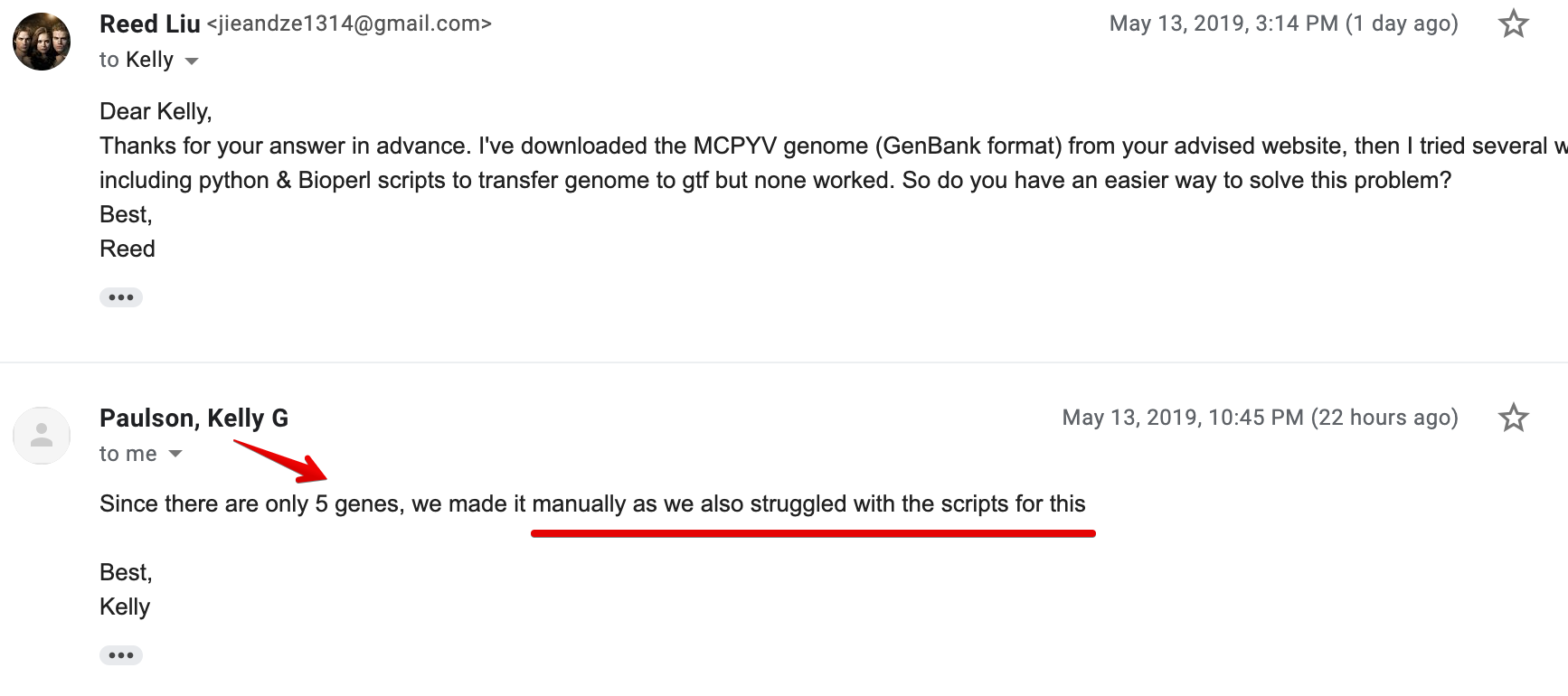
本以为人家不会给回信息,因为问的问题好像有点和他们文章没什么关系了,但他真的回信了并且很负责告诉我:"他也不会!",解决办法就是:手动构建一个
其实这个事情可以告诉我们,复现文章中遇到问题时,直接和原作者沟通是最有效的方式(这是我第一次和一作直接进行交流)。之前的时间都是自己在摸不着头脑地进行各种尝试,然后找代码,结果都不可以成功
先把基因组序列下载下来
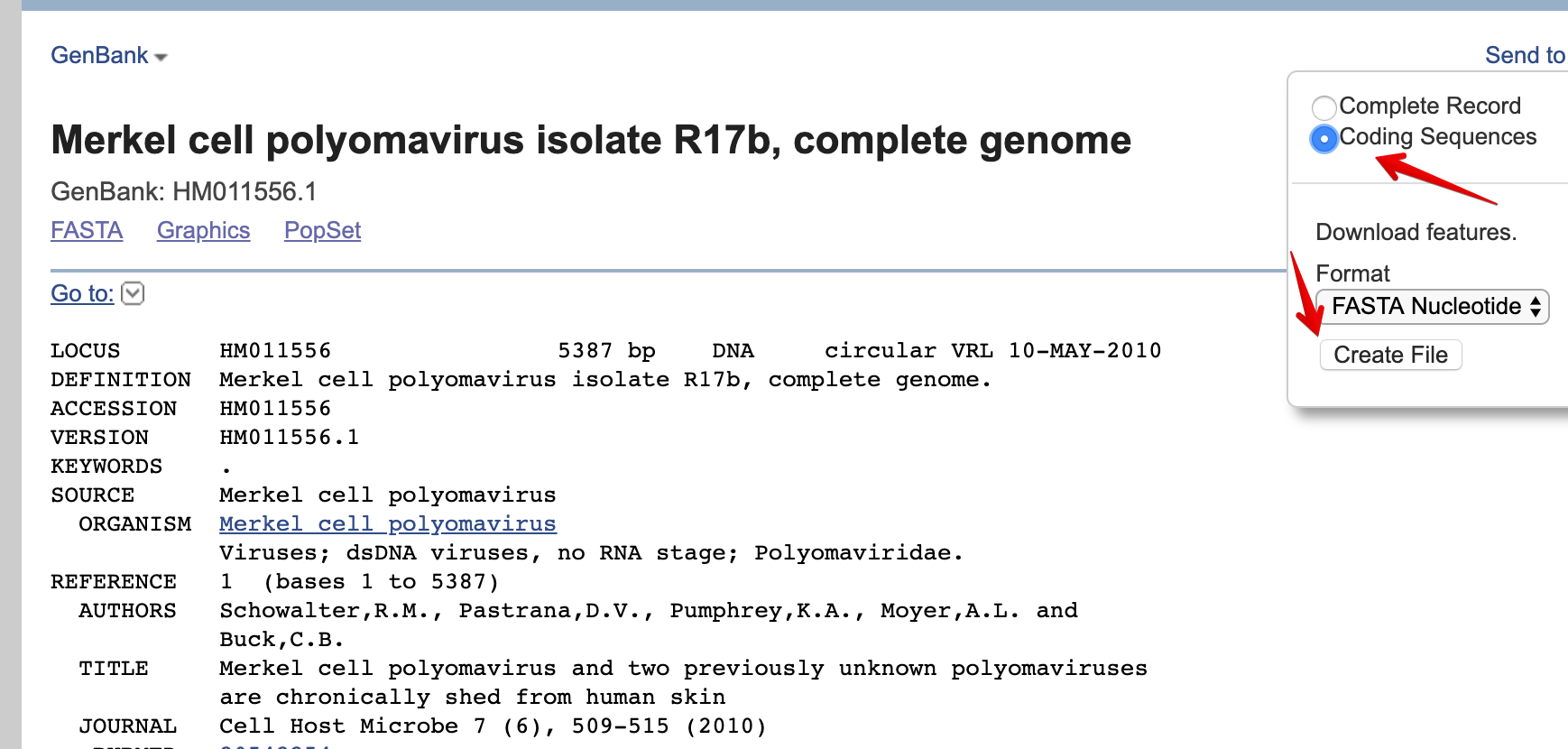
然后根据genbank的序列信息,造出5个CDS的注释信息
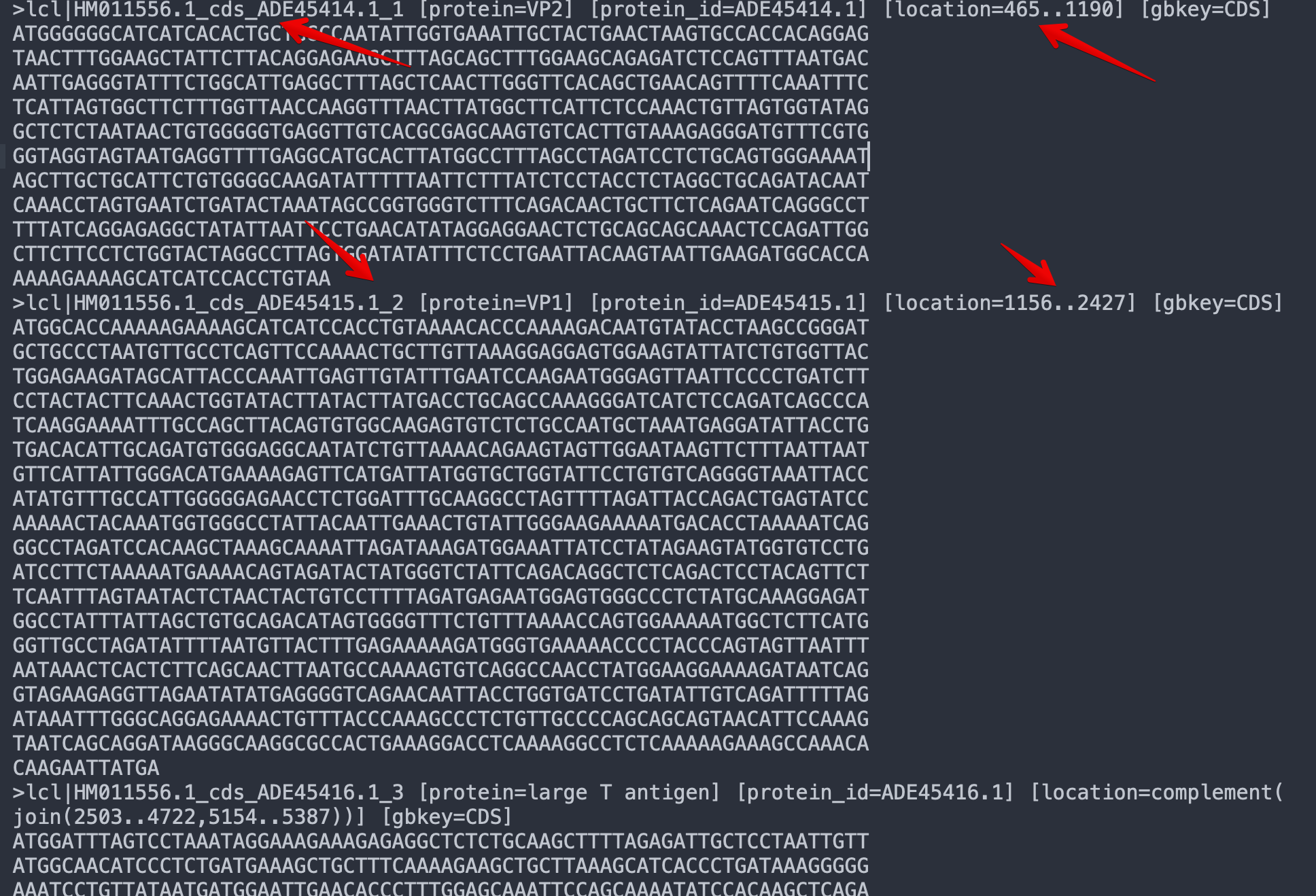
基本就按照这个样子来就好,不需要很复杂
需要注意的是,cellranger只能识别exon,所以我们也要这样设计
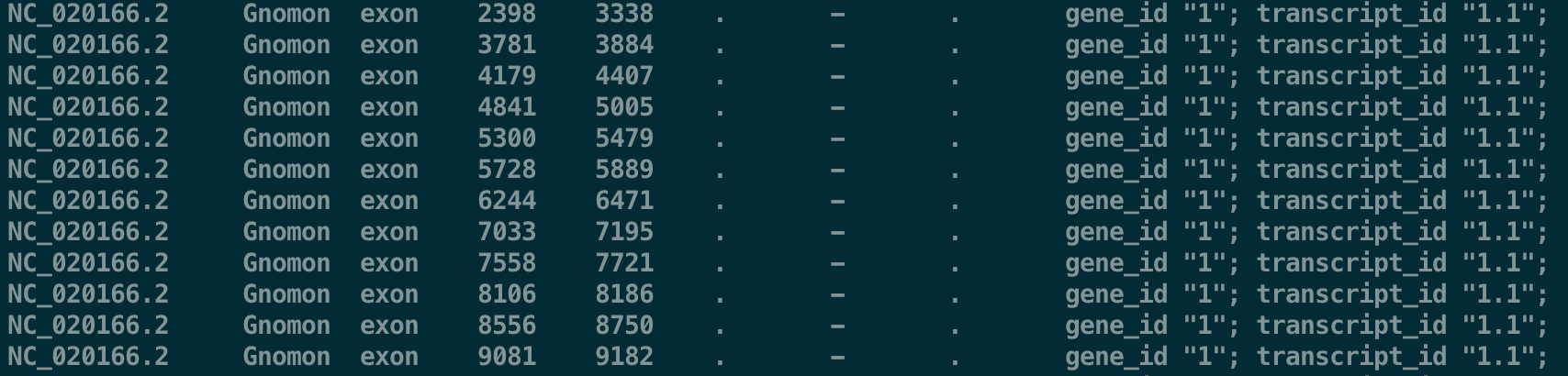
- # 每一行有9列tab分隔信息
- # 第一列:Chromosome 指定基因组上染色体或contig位置
- # 第二列:Source 这个用处不大
- # 第三列:Feature CellRanger软件只取exon的部分
- # 第四列:Start 起始位点(1-based)
- # 第五列:End 终止位点(1-based)
- # 第六列:Score 这个用处不大,建议用"."表示
- # 第七列:Strand feature信息在基因组的+或-链
- # 第八列:Frame 用处不大,建议“.”
- # 第九列:分号分隔的键值对,重点是transcript_id 和gene_id。gene_name可选
- ADE45414.1_1 Gnomon exon 465 1190 . - . gene_id "1"; transcript_id "1.1";
- ADE45415.1_2 Gnomon exon 1156 2427 . - . gene_id "1"; transcript_id "1.1";
- ADE45416.1_3 Gnomon exon 2503 4722 . - . gene_id "1"; transcript_id "1.1";
- ADE45416.1_3 Gnomon exon 5154 5387 . - . gene_id "1"; transcript_id "1.1";
- ADE45417.1_4 Gnomon exon 4827 5387 . - . gene_id "1"; transcript_id "1.1";
但是自己构建时,一定要注意使用tab分隔,即使看上去像也不可信,检查的方法有两个:
一个是直接运行cellranger mkgtf 看是否报错,可能会提示:GTF的行数不对;
另一个是直接检查:awk -F 't' '{print NF}' mcv.gtf,如果显示1,那么说明没有tab分隔,而是用的多个空格,利用sed可以把这些空格替换成tab:
sed 's/ + /t/g' inputfile > outputfile