

最近为了了解下GSEA的算法的整体思路以及软件一些主要参数的具体含义,从头看了下其R代码,由于其R代码是作者在软件早期写的,其功能并不是很完善,但是其核心的思路还是在代码中体现出来了(其实主要还是GSEA的JAVA版看不懂。。)
下面结合作者发表的文献、官方手册以及R代码,对一些有意义的参数和一些指标做个简单的记录,也算一次看代码后的小结,如果可能的话,也想从头写一个简化版的GSEA的R代码(主要发现官方的R代码写的年代好早之前了,代码写的有点啰嗦~~)以及其出图的代码
理解GSEA参数
- Signal2Noise参数这个是GSEA默认的gene ranking方法
Signal2Noise (default) uses the difference of means scaled by the standard deviation. Note: You must have at least three samples for each phenotype to use this metric.
公式很简单:
- (μa-μb)/(σa+σb)
- > where μ is the mean and σ is the standard deviation; σ has a minimum value of .2 * absolute(μ), where μ=0 is adjusted to μ=1. The larger the signal-to-noise ratio, the larger the differences of the means (scaled by the standard deviations); that is, the more distinct the gene expression is in each phenotype and the more the gene acts as a “class marker.”
- 公式中的a和b分别代表着两组样本,从代码中也能看到其根据cls文件的信息,确定要分组样本,然后根据表达矩阵计算每组样本的均值和方差,最后计算signal to noise scores;最后作者也做了一点调整,其将方差的最小值设定为均值绝对值的0.2倍,也就是说如果方差小于绝对均值的0.2倍,则将0.2倍绝对均值赋值于方差
- 除了Signal2Noise方法外,GSEA的java版还有其他8种方法可供选择,分别是`tTest`,`Cosine`,`Euclidean`,`Manhatten`,`Person`,`Ratio_of_Classes`,`Diff_of_Classes`,`log2_Ratio_of_Classes`;这里几种应该中应该有些看名字能猜出点啥,其实GSEA官方手册都列出其各个方法的公司,翻一下就知道了[http://software.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html](http://software.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html)
- 对于Max Size和Min Size参数这个参数其实也蛮有意思的,有时会发现你的GSEA结果为啥富集的通路很少,一些关心的通路则根本没在结果中出现,一种原因就是其根本没参与GSEA的计算,可以先看看这段话:
When you run the gene set enrichment analysis, the GSEA software automatically normalizes the enrichment scores for variation in gene set size, as described in GSEA Statistics. Nevertheless, the normalization is not very accurate for extremely small or extremely large gene sets. For example, for gene sets with fewer than 10 genes, just 2 or 3 genes can generate significant results. Therefore, by default, GSEA ignores gene sets that contain fewer than 25 genes or more than 500 genes; defaults that are appropriate for datasets with 10,000 to 20,000 features. To change these default values, use the Max Size and Min Size parameters on the Run GSEA Page; however, keep in mind the possibility of inflated scorings for very small gene sets and inaccurate normalization for large ones
一个gene set会有对应的N个gene symbol,要么作者认为,如果当你的表达矩阵中能跟gene set中的gene list相比匹配的基因过多或者过少都是不合理的,会在对ES标准化时造成偏差;当你的表达矩阵的gene数目在10000-20000之间时,其默认参数是最小数目为25(GSEA软件里是15),最大数目是500
- ES值(enrichment scores)ES值应该说是GSEA最有特色的一个数值,其代表了GSEA整个分析思路的核心
For a randomly distributed S, ES(S) will be relatively small, but if it is concentrated at the top or bottom of the list, or otherwise nonrandomly distributed, then ES(S) will be correspondingly high.
作者认为当其表达矩阵的gene list在gene sets中是随机分布的话,那么最终的ES值是相对较小的;当是非随机分布时,则对应的ES值是相对较大的。这在置换检验中就很好理解了
ES计算公式如下:
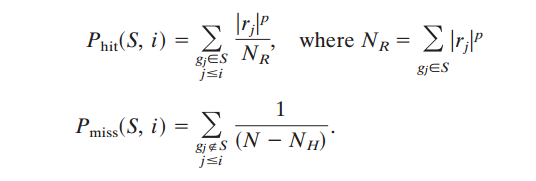
ES值相当于Phit - Pmiss差值的最大累计值(绝对值);N就是总gene list的数目;NH则是Hit gene list的数目;NR在不同情况下含义有点差别,比如在Signal2Noise下,其就是Hits-Gene在Ranking gene所对应的signal to noise scores总和,从图片上就是中间竖直黑线所对应值的求和;所以简单的说,以Signal2Noise为例,Phit值就是Hit gene的scores除以所有Hit gene的scores值的和
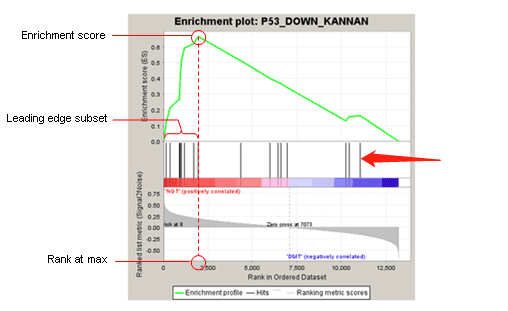
其是以FC ranking为例的,以图+表格的形式的表述了ES值的产生,个人觉得这张图将的更加易懂

- Permutation type参数选择置换形式,主要用于计算enrichment score(ES)的显著性,可选两种形式:Phenotype和Gene_set,简单的理解就是要么选择置换样本,要么选择置换gene sets。
需要注意的是,当选择Phenotype时,计算ES值需要用到ranking scores,所以每随机置换一次,都会ranking一次gene list,然后计算一个ES值,然后根据这一系列的ES值形成一个null distribution,然后将observe或者说actual的ES值与上述一系列ES比较,最终获得一个显著性P值,作者建议当每组样本数大于7个时使用
当选择Gene_set顾名思义则是随机gene sets,也是跟上面一样,形成一系列的ES值,然后与actual的ES值比较,获得P值(gene sets代码未看,其R代码里面没有这步。。。所以具体思路不太清楚),但是作者也做了解释:由于在创建随机gene sets时,其不像随机Phenotype那样ranking和求ES的gene list是同步的(这句话我也解释不清楚)
- GSEA的Multiple Hypothesis Testing从GSEA的结果中我们可看到,除了从置换检验获得的NOM p-val外,还有其他多重假设检验的结果,如:FDR q-val和FWER p-val
NOM p-val值,计算很简单,如果是actual ES是正值,则只需要计算actual ES大于随机ES(正值)的个数在总ES(正值)中的比例;如果是负值,则计算actual ES小于随机ES(负值)的个数在总ES(负值)中的比例
FWER p-val值,首先需要对ES值进行标准化,对每个gene set计算NES值,计算也很简单,就是将ES值除以所有随机样本的ES的均值。接着在NES值的基础上,获得每次随机的各个gene sets中NES的最大值(正负NES值分开计算),最后将actual NES值与上述随机的NES最大值进行比较,这里就跟NOM p-val一样了,计算比例
FDR q-val值,先对每个actual的gene sets的NES值降序,接着当NES值大于0时,计算每个gene sets下随机样本中NES值大于0的个数,再计算随机NES值大于actual NES值的个数占总NES大于0的数目的比例;当NES为负数时也一样;接着计算上述比例的平均数以及中位数;然后以随机样本的NES平均数/中位数与actual样本的NES平均数/中位数的比值作为FDR.mean/FDR.median;最后输出的FDR q-val则是FDR.mean
虽然看代码时理解的比较清楚,但是写出来后就发现有点乱了,所以相比看文章,看是倾向于看代码,比如文章对于FDR q-val是这样的:
Create a histogram of all NES(S, π) over all S and π. Use this null distribution to compute an FDR q value, for a given NES(S) = NES* ≥ 0. The FDR is the ratio of the percentage of all (S, π) with NES(S, π) ≥ 0, whose NES(S, π) ≥ NES, divided by the percentage of observed S with NES(S) ≥ 0, whose NES(S) ≥ NES, and similarly if NES(S) = NES* ≤ 0
GSEA服务器版
除了了解上述GSEA界面版中常见的参数外,其实GSEA的java服务器版的参数也要略微了解下,因为有时当你需要批量处理一系列数据时,或者本地电脑内存不够时(因为GSEA桌面版总高是8G内存,因此如果你的数据量很大时,其是跑不动的),GSEA的java服务器版就是较佳的选择
使用前先看下官方手册http://software.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html?_Other_Ways_to
模仿着给出例子的命令行(选个简单的,其他都是默认参数)
- java – Xmx1024m -cp /xchip/projects/xtools/gsea2.jar xtools.gsea.Gsea -res test.gct -cls test.cls -gmx test.gmx -collapse false
如果对于其他参数有要求,可以查看下GSEAParameters.txt,然后依据实际情况修改下,然后用–param_file参数导入
比如我用的命令行:
- java -Xmx10240m -cp ./gsea-3.0.jar xtools.gsea.Gsea -res cancer.txt -cls group.cls --gmx c2.cp.v6.1.symbols.gmt -chip ENSEMBL_human_gene.chip -collapse true -plot_top_x 50 -out gsea_cancer
但一些细节上的分析可能还是GSEA桌面版做的比较好