

1. 如何选择参考基因组?——李恒2017年年底博客
1.1 三种选择
- 如果比对到GRCh37/hg19,使用:
ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/reference/human_g1k_v37.fasta.gz
- 如果比对到GRCh37/hg19,并且认为包含
decoy序列能够更准确地进行变异检测,使用:
ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz
- 如果比对到GRCh38/hg38,使用:
ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz
1.2 其他版本的GRCh37/GRCh38有什么问题?
- 包含ALT contig序列
ALT contigs are large variations with very long flanking sequences nearly identical to the primary human assembly. Most read mappers will give mapping quality zero to reads mapped in the flanking sequences. This will reduce the sensitivity of variant calling and many other analyses. You can resolve this issue with an ALT-aware mapper, but no mainstream variant callers or other tools can take the advantage of ALT-aware mapping.
个人理解:上面说的是一个多比对问题。新版的bwa可以处理ALT-aware的比对
ALT contig是什么?
To better capture variation in the human genome across the world;
it(hg38最初版) contains more copies of some loci than hg19(最初版).
Patches(补丁)是什么?
patch releases do not change any previously existing sequences; they simply add new sequences for fix patches or alternate haplotypes that correspond to specific regions of the main chromosome sequences. For most users, the patches are unlikely to make a difference and may complicate the analysis as they introduce more duplication. (patches和ALT contigs的概念有些重合)Alternate loci scaffolds: - a scaffold that provides an alternate representation of a locus found in the primary assembly. These sequences do not represent a complete chromosome sequence although there is no hard limit on the size of the alternate locus; currently these are less than 1 Mb. These could either be NOVEL patch sequences, added through patch releases, or present in the initial assembly release. - format: chr{chromosome number or name}_{sequence_accession}v{sequence_version}_alt - e.g. chr6_GL000250v2_alt
所以ALT contig序列是为了反映人群多态性的一段替补序列,和原染色体位置对应的序列之间有一定的差异。放在ref中的隐患是人为增加了重复序列。
- 用很长的N间隔这些ALT contig序列增加了不必要的ref的size
- 存在多处定位的参考序列比如X染色体上的一些区域在Y染色体中也有(pseudoautosomal region, PAR)。在标准的流程中,这些区域是没法进行变异检测的(怎么理解?上面第一点也提到了)。正确的做法是将Y染色体中的这类区域进行mask。
- 没有使用rCRS的线粒体序列rCRS在群体遗传学中广泛用到,但是官方的GRCh37的线粒体序列比rCRS长2bp,构建线粒体的系统发生树应该使用rCRS。GRCh38使用的是rCRS。
- 将模棱两可的碱基编码为Nhttp://biocorp.ca/IUB.php
- 使用accession numbers而不是chrXXX作为染色体名称
- 不包含未精确定位的contig序列
hg19/chromFa.tar.gz from UCSC: 1, 3, 4 and 5.
hg38/hg38.fa.gz from UCSC: 1, 3 and 5.
GCA_000001405.15_GRCh38_genomic.fna.gz from NCBI: 1, 3, 5 and 6.
Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz from EnsEMBL: 3.
Homo_sapiens.GRCh38.dna.toplevel.fa.gz from EnsEMBL: 1, 2 and 3.
共性问题是3
翻译参考[1]
1.3 针对`1.1`前两条的说明
human_g1k_v37.fasta包含了人的24条染色体水平的序列,线粒体序列,以及没有定位的contig序列。
hs37d5.fa包含了以上,以及NC_007605、hs37d5:
NC_007605是人疱疹病毒的序列,Human herpesvirus 4 type 1,也称EBV;
hs37d5就是decoy序列,由很多短序列间隔一定数量的N组成。
这些短序列是什么?有什么作用?
这些短序列可以简单认为是来源于人但是ref里面不包含(组装技术的局限)。怎么找到的?对比已有的克隆序列和ref序列确定的。参考基因组并不完整,总有一些区域不包含在已有的参考基因组中,这种情况下,来源于这些区域的reads要么比对不上,要么错误地比对到其他地方(造成假阳性)。如果能把那些已经确定的但是ref没有的序列加上去,就能有所改善。
EBV序列和decoy序列的作用类似,都是尽可能让reads比对到它真实的地方。只不过EBV序列不属于human genome,但细胞里面可能含有,提取DNA测序可能就测到了。
参考[2]
1.4 个人搜集
Ensembl
可以下到最新版
ftp://ftp.ensembl.org/pub/release-98/fasta/homo_sapiens/dna/
ftp://ftp.ensembl.org/pub/release-98/gtf/homo_sapiens/
GATK
https://software.broadinstitute.org/gatk/download/bundle
包括SNP, InDel这类为变异检测提供参考的文件。
现在好像进不了
NCBI
最新版
ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/405/GCF_000001405.39_GRCh38.p13
2. 认识GRCh38/hg38
- 包含alternate contigs,能反映群体的单体型比如HLA alternate,这段序列多态性非常高,一个ref不一定具有代表性。
- The GRCh38 analysis set hard-masks regions and provides decoy contigs for optimal read mapping(见下文NCBI部分)
- The challenge alternate contigs presents is a familiar one
- Latest versions of BWA-MEM handle GRCh38 alternate contig mappings这一点是说最新版的bwa可以支持比对到这类ALT区域了,针对李恒说的
1.2第一点。 - Alt-handling requires the SAM format ALT index file
- New Tutorial#8017 shows how to map to GRCh38 with alt-handling and then some
- Simulate read mapping for your favorite alternate haplotype
- Our production workflow for single sample variant calling on GRCh38 is public and uses shiny new features
- Finally, there is no better time than now to start learning WDL
参考[3]
- Inclusion of model centromere sequences
3. Masked, soft-masked and unmasked genomes
Ensembl上的参考基因组有以上三种形式,分别表示
- 将重复区域和低复杂度区域替换为N信息缺失
- 将重复区域和低复杂度区域替换为小写和第三种没什么区别,因为软件大小写一致对待。But it should be noted that most of the alignment tools do not take into account soft-masked regions, for example BWA, tophat, bowtie2 tools always use all bases in alignment weather they are in lowercase nucleotide or not.
- 不做处理推荐用这种
参考[4]
4. UCSC
http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/
以这个hg38文件夹为例,在这个文件夹中有几个小文件夹:
4.1 initial/
GRCh38最初的版本,包含the original alternate sequences (261) and no fix sequences,和NCBI上的GCA_000001405.15有区别吗?
hg38.chromFa.tar.gz - The assembly sequence in one file per chromosome.
Repeats from RepeatMasker and Tandem Repeats Finder (with period of 12 or less) are shown in lower case; non-repeating sequence is shown in upper case.
hg38.fa.gz - "Soft-masked" assembly sequence in one file.
Repeats from RepeatMasker and Tandem Repeats Finder (with period of 12 or less) are shown in lower case; non-repeating sequence is shown in upper case.
4.2 latest/
GRCh38最新的版本,注意这里的最新是UCSC的最新,可能NCBI更新更快。比如此时UCSC最新是GRCh38.p12,但NCBI已经到GRCh38.p13了。
还有几个重要文件:
4.3 mrna.fa.gz
Human mRNA from GenBank. This sequence data is updated regularily via automatic GenBank updates.
4.4 refMrna.fa.gz
RefSeq mRNA from the same species as the genome. This sequence data is updated regularily via automatic GenBank updates.
4.3和4.4可能是一样的
4.5 标准的基因组序列文件
For most users, this will be the file "latest/hg38.fa.gz" in this directory.
http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/latest/
4.6 分析集序列文件
if you need a genome file for alignment or variant calling, please read the section "Analysis set" below.
http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/analysisSet/

下面两个较上面多了alt-scaffolds;2,4是soft-masked的;第2个文件同the NCBI file GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz
4.7 与NCBI ENSEMBL的比较
主要染色体的序列在几个数据库中是一样的,但染色体名称不同。
"chr1" at UCSC, "NC_000001.11" at NCBI, and "1" at the ENSEMBL;
soft-masked的碱基不完全一样,因为三者运行Repeatmasker设置参数不一样
5. NCBI
最新版是GRCh38.p13,GCF_000001405.39
https://www.ncbi.nlm.nih.gov/genome/guide/human/
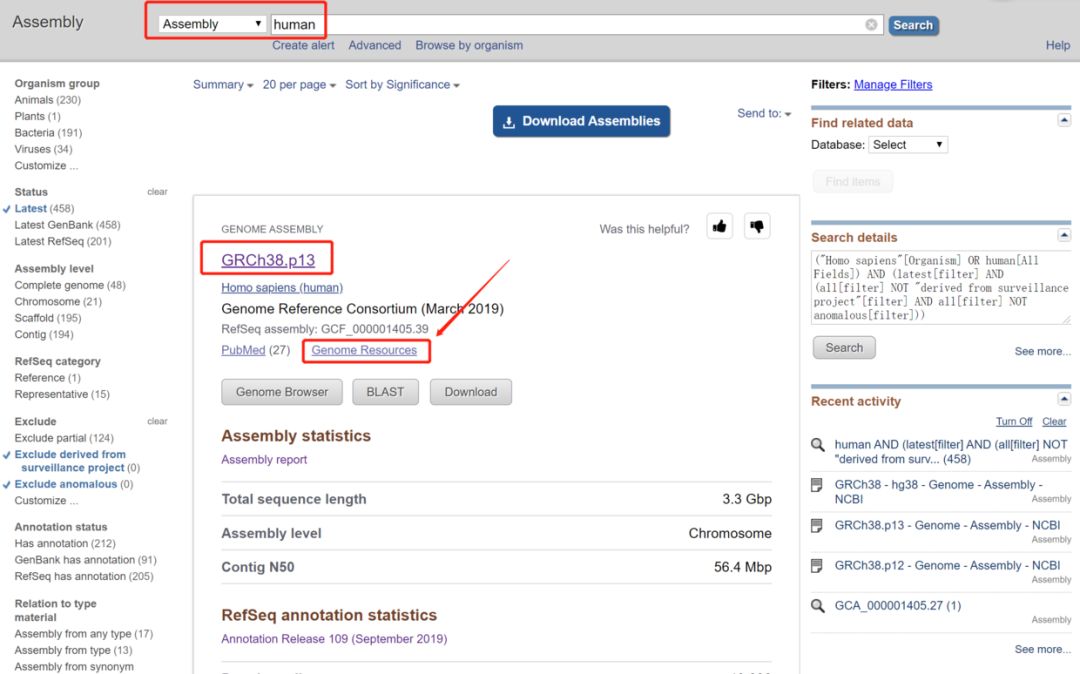
同样的,包含主要染色体,未定位的scaffold序列,patch和alt contig序列。但是patch和alt contig序列是直接追加的,没有在两端加N,因此解压出来3.1G左右。
gff3文件信息很丰富,条目包括:CDS, enhancer, exon, gene, lnc_RNA, miRNA, rRNA, scRNA, snRNA, snoRNA, tRNA, TATA_box等等。
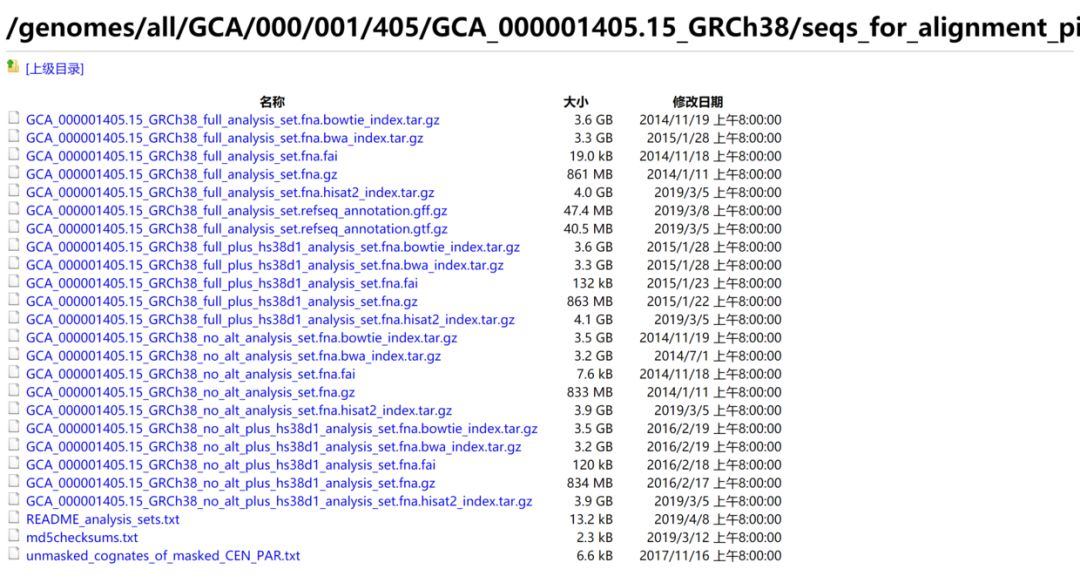
下面那个红框是专门为分析流程准备的文件,包含bwa, hisat2, bowtie等的索引,也就是分析集文件,如下:
6. ensembl
官网:http://asia.ensembl.org/index.html
ftp://ftp.ensembl.org/pub/release-98/gff3/homo_sapiens/
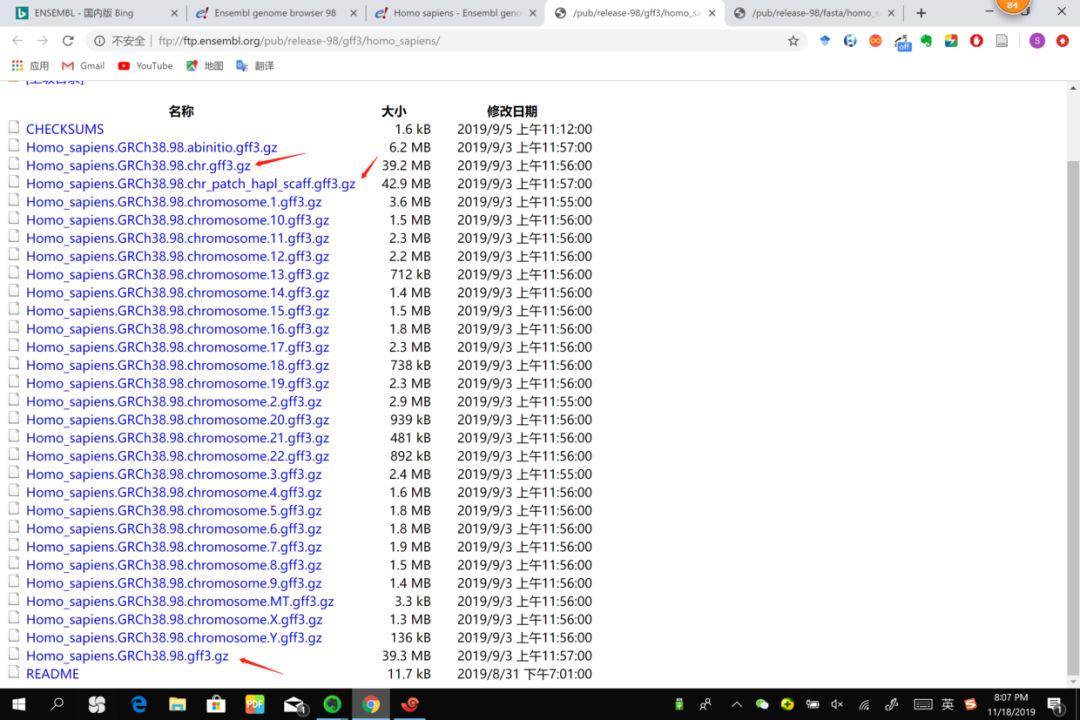
这几个有什么区别?
- 98.chr.gtf.gz 只包含主要染色体(有MT)的注释
- 98.gtf.gz 包含主要染色体以及一些没有定位的染色体片段上的注释信息
- 98.chr_patch_hapl_scaff.gtf.gz 除了上面的,还有patches和ALT contig的注释
条目包含:CDS, exon, 5'-UTR, 3'-UTR, stop_codon等等常见的条目。
ftp://ftp.ensembl.org/pub/release-98/fasta/homo_sapiens/dna/
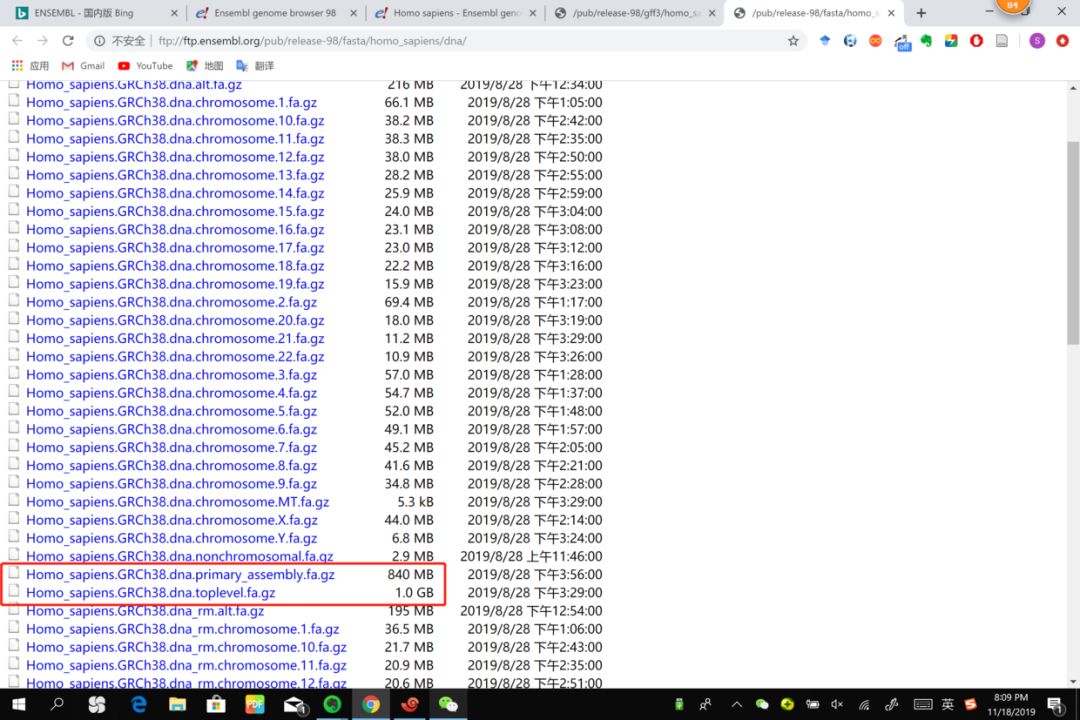
6.1 toplevel & primary_assembly
toplevel
This includes chromsomes, regions not assembled into chromosomes and N padded haplotype/patch regions.
这个文件解压出来有60G,远大于3G,增加了很多无用的N。比如一个MHC基因座(位于chr6)的多态性序列,两端会增加很多N,直到序列约等于chr6的长度,再添加到ref中。
primary_assembly
Primary assembly contains all toplevel sequence regions excluding haplotypes and patches. This file is best used for performing sequence similarity searches where patch and haplotype sequences would confuse analysis. If the primary assembly file is not present, that indicates that there are no haplotype/patch regions, and the 'toplevel' file is equivalent.
7. 思考
尽管在1.中,李恒老师列出了一些ref的不足,但实际分析中选不同的参考基因组影响不太大(他的博客也提到了这一点)。另外,NCBI和ENSEMBL更新快于UCSC。
尽管patches和ALT contigs会不断更新,GRCh的次版本(比如GRCh38.p13)也会随之更新,但主要染色体的gff/gtf应该是不变的,因此不用担心换了ref的次版本而找不到注释文件,因为大多数时候我们只看主要染色体上的注释。
hg38提及decoy比较少,可能组装过程中已经整合进去了一部分,另外在NCBI分析集中提到了decoy(见上文)
7.1 需要探究的问题?
在top level中,patch和ALT contig是如何存放的?
每一个patch或者alt contig序列都是前后增加了很多N,然后追加到primary中。
NCBI和ENSEMBL是否都含有ncRNA的注释?
NCBI有,ENSEMBL没有。
或者这样问:它们的gtf有什么区别?NCBI的条目更多。
另外:NCBI上面的染色体命名不好认,基因、转录本的ID没有ENSEMBL全。
gencode的注释和上文的gtf有关吗?
- GENCODE的最新版(Release 32)的注释就是ENSEMBL最新版的默认注释集(GRCh38.p13)。另外,GENCODE还提供了lncRNA, tRNA的gtf/gff文件,这个ENSEMBL上面没有。
- 而USCS将GENCODE作为一个子集track:
http://genome.cse.ucsc.edu/cgi-bin/hgTables
- NCBI的注释是它自己的,叫“NCBI RefSeq”。在UCSC中也能下载这个子集track的gff/gtf。
8. reference
[1] https://lh3.github.io/2017/11/13/which-human-reference-genome-to-use
[2] https://www.biostars.org/p/73100/
[3] https://software.broadinstitute.org/gatk/blog?id=8180
[4] https://genestack.com/blog/2016/07/12/choosing-a-reference-genome/