

简单介绍F-statistics 和 Fst
1. 概念
在群体遗传学中,F-statistics(Fixation indices)是衡量种群中基因型实际频率是否偏离遗传平衡(哈温平衡)理论比例的指标。F-statistics的概念由美国遗传学家Wright在文章中提出,当时他对牛的近亲繁殖很感兴趣,然而,由于完全显性优势导致显性纯合子和杂合子控制的表型相同,直到20世纪60年代以来分子遗传学的出现,遗传学家们才得以测量群体中的杂合度。
F-statistics也可以被认为是在分层群体(如生活在高原的高海拔人群和生活在平原的低海拔人群)中不同亚群间基因相关性的度量。这种相关性 (也可以理解为genetic distance) 会受到进化事件的影响,如遗传漂变,瓶颈效应,遗传搭载效应(有利突变附近位点由于连锁随着主效应位点一起频率升高并逐渐在群体中固定下来),突变,基因交流,近亲繁殖,自然选择等。这些进化事件导致的改变都会反映在等位基因频率和单倍型频率上,不过目前大多算法都是针对二等位基因设计,多等位基因的计算效果还有待进一步评估。
在这里我们先定义一些符号:
O(f(Aa))--实际观察杂合子频率,E(f(Aa))--期望杂合子频率,N表示数目。
在群体结构为两个层次的群体中:一个层次为个体(I)到亚群(S),另一个从亚群(S)到总群体(T).
HI 表示总群体所有个体实际观察到的杂合子频率平均值;
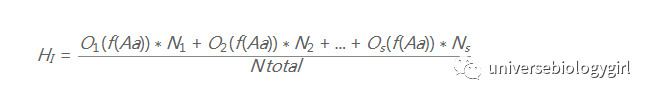
HS 表示亚群在哈温平衡下杂合子期望频率:
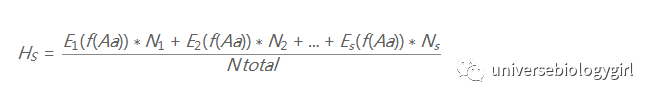
HT 表示总群体在哈温平衡下杂合子期望频率;
![]()
FIT 表示个体(I)相对于总群体(T)的近交系数,即总群体的平均近交系数;
![]()
FIS 表示个体(I)相对于亚群(S)的近交系数,即亚群的平均近交系数;
![]()
FST 表示亚群(S)相对于总群体(T)的近交系数,即有亲缘关系亚群间的平均近交系数。
![]()
FIT, FIS, FST之间的关系可以表示为 (1-FIS) = (1-FIS) (1-FST) .
在哈温平衡下,近交系数计算公式如下:

并且预期杂合频率为: E (f(Aa)) = 2pq
2. 举例
上面讲了这么多公式,下面通过一个例子来加深理解。
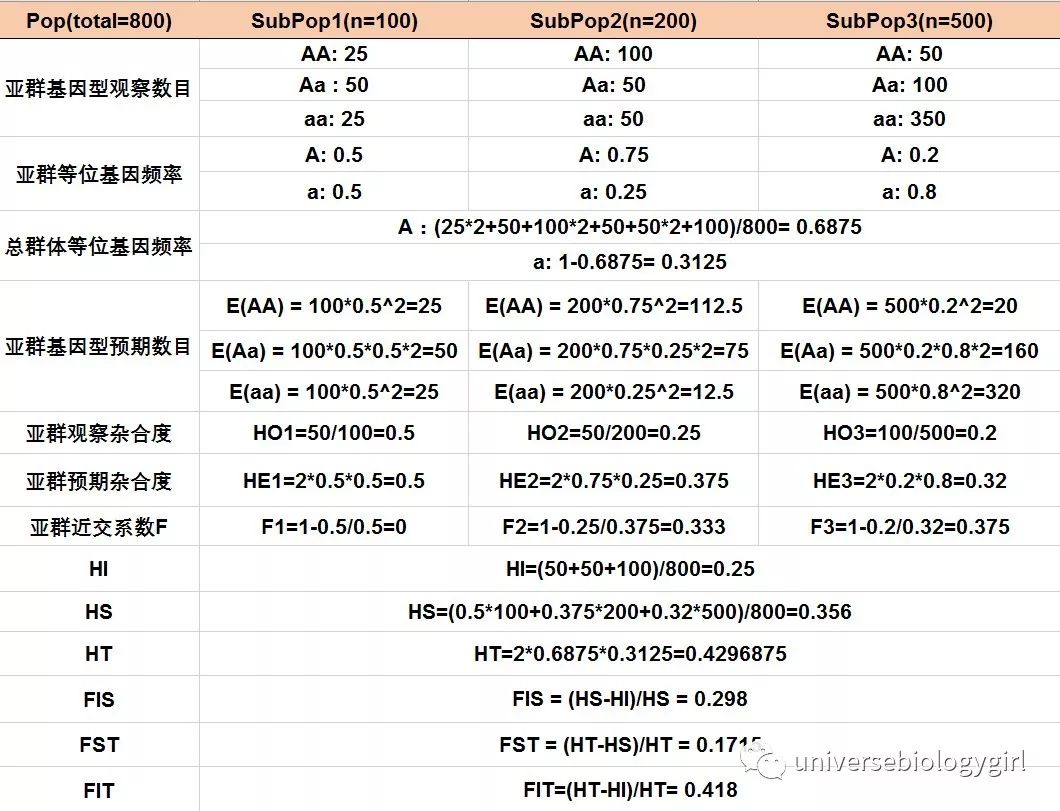
FST的取值范围为[0,1], 数值越大,表明等位基因在各自亚群中越固定(频率越高),群体间分化程度越大。
实战
1. 计算FST
在实际项目中,样本很难完全代表总体,上述各统计量也很难得到,因此出现了很多FST计算方法,现在应用最广泛的是Weir & Cockerman(1984)提出的方法。
数据上采用Gou et al (2014, Genome Reserch) 中的数据。
| Breed | Altitude | Size |
|---|---|---|
| Tibetan Mastiff (TM) | 4500m | 10 |
| Diqing indigenous dog (DQ) | 3500m | 10 |
| Lijiang indigenous dog (LJ) | 2500m | 10 |
| Kunming dog (KM) | 1800m | 10 |
| German Shepherd (GS) | 1800m | 10 |
| Yingjiang indigenous dog (YJ) | 800m | 10 |
进行质控,剔除高缺失率(--geno 0.05)和极低等位基因频率( --maf 0.01 )的SNP
- #!/bin/bash
- plink=/software/biosoft/software/plink/plink
- $plink --vcf 60dog.vcf --geno 0.05 --maf 0.01 --dog --recode vcf-iid --out 60dog_QC
这里以TM和YJ为例,计算FST,因为逐个位点计算FST时,可能会出现FST值很高的假阳性信号(中性选择导致),所以这里考虑到搭载效应同时计算了滑窗FST,二者可以对照着看。TM_4500.txt为TM样本ID文件,一行一个ID,YJ_800.txt同左。
- #!/bin/bash
- vcftools=/software/biosoft/software/vcftools_0.1.13/bin/vcftools
- # 单位点计算Fst
- $vcftools --vcf 60dog_QC.vcf --weir-fst-pop YJ_800.txt --weir-fst-pop TM_4500.txt --out TM_4500vsYJ_800_single_locus
- # 窗口
- $vcftools --vcf 60dog_QC.vcf --weir-fst-pop YJ_800.txt --weir-fst-pop TM_4500.txt --fst-window-size 20000 --fst-window-step 5000 --out TM_4500vsYJ_800_20k_5k
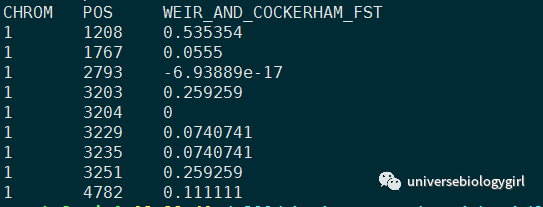
这里可以来看下结果文件
单位点:
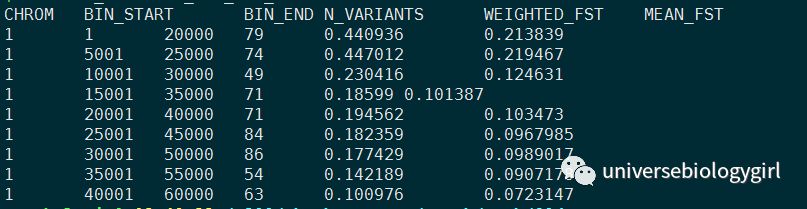
2. 可视化
这里使用R包qqman完成曼哈顿图可视化,以窗口结果为例。
先处理数据文件格式:
- sed "1d" TM_4500vsYJ_800_20k_5k.windowed.weir.fst|awk '{if($5<0)print $1"\t"$2"\t0";else print $1"\t"$2"\t"$5}' > TM_4500vsYJ_800_20k_5k_plot.txt
可视化脚本代码
- #!/usr/bin/env Rscript
- rm(list=ls())
- ### 加载要使用的R包
- library(qqman)
- library(Cairo)
- Fstfile<-read.table("TM_4500vsYJ_800_20k_5k_plot.txt", header=F, stringsAsFactors=F)
- SNP<-paste(Fstfile[,1],Fstfile[,2],sep = ":")
- Fstfile=cbind(SNP,Fstfile)
- colnames(Fstfile)<-c("SNP","CHR","POS","Fst")
- outfile<-"TM_4500vsYJ_800_20k_5k"
- filePNG<-paste(outfile,"manhattan.png",sep=".")
- CairoPNG(file=filePNG,width=1500,height=500)
- colorset<-c("#FF0000","#FFD700","#2E8B57","#7FFFAA","#6495ED","#0000FF","#FF00FF")
- ### 调用qqman,画出全部染色体
- manhattan(Fstfile),chr="CHR",bp="POS",p="Fst",snp="SNP", col=colorset,logp=F,suggestiveline = F, genomewideline = F,ylab="Fst",ylim=c(0,1),font.lab=4,cex.lab=1.2,main="TMvsYJ",cex=0.8)
- ### 只绘制单条染色体
- if (FALSE){
- manhattan(subset(Fstfile,CHR=="10"),chr="CHR",bp="POS",p="Fst",snp="SNP", col=colorset,logp=F,suggestiveline = F, genomewideline = F,ylab="Fst",ylim=c(0,1),font.lab=4,cex.lab=1.2,"TMvsYJ_chr10",cex=0.8)
- }
- dev.off()
- print("==================manhattan plot====================")
可视化结果展示
全部染色体

10号染色体

小结
本篇文章介绍了FST的计算过程,实战过程和可视化。FST是目前最基础也是最常用的检测自然选择的方法,但是由于FST会显示出所有分化程度较高的位点和基因,所以有时候不是十分贴合我们的目前性状或环境因素,后续还需要结合其他分析综合判断。
参考文献:
https://en.wikipedia.org/wiki/F-statistics
https://en.wikipedia.org/wiki/Fixation_index
Weir, B. S., and C. Clark Cockerham. “Estimating F-Statistics for the Analysis of Population Structure.” Evolution, vol. 38, no. 6, 1984, pp. 1358–1370. JSTOR, www.jstor.org/stable/2408641.