大家对ROC曲线都很熟悉,从方法的特异性和灵敏度出发反应一个方法的准确度。但是,在临床的应用中,往往仅通过以上标准得到的准确度是不可靠的。故早在2006年纪念斯隆-凯特琳癌症中心AndrewVickers博士等人研究出另外一个新的评估方法,叫决策曲线分析法(Decision Curve Analysis,DCA)。今天我们就来介绍下在R语言中如何实现决策曲线分析方法。
首先我们 还是找到一个DCA的包DecisionCurve。当然现在这个包还未在官方库中出现,目前安装需要先载入devtool包,然后利用gihub的资源进行包的安装具体代码如下:
- library(devtools)
- install_github("mdbrown/DecisionCurve")
安装完成界面如下图:

还未等你回味安装的艰辛,其实我们发现其安装后名字也已经不再是DecisionCurve而改成了rmda。那么我们就来看下其函数构成:
1. decision_curve

参数说明:
formula主要是因变量和自变量的变量名称设置,中间用~区分开。当然在这里因变量都是二进制数据。
family =binomial(link = ‘logit’)是使用logistic回归来拟合模型。threshold设置横坐标阈概率的范围,一般是0-1;但如果有某种具体情况,大家一致认为Pt达到某个值以上,比如40%,则必须采取干预措施,那么0.4以后的研究就没什么意义了,可以设为0-0.4。by是指每隔多少距离计算一个数据点。
2. summary.decision_curve 等同于summary列出评估模型的所有内容
3. plot_decision_curve 绘制以上生成的决策曲线
4. plot_clinical_impact 绘制每个计算点的样本数量。主要是查看真实分布和模型预测分布之间的差异或者说是否一致。

5. plot_roc_components绘制ROC的假阳性和真阳性的概率分布。
6. cv_decision_curve以交叉验证方式构建评估模型。

以上函数构成了DCA包的主要函数,接下来我们看下一些实例,进一步了解其构建过程及意义。所有的数据依据代码都是运行的包的实例,在这里只是做相关的解说。
首先,我们看下简单的模型构建:
- data(dcaData)baseline.model <- decision_curve(Cancer~Age + Female + Smokes, data = dcaData, thresholds = seq(0, .4, by = .01), study.design = 'cohort', bootstraps = 10)
- full.model <- decision_curve(Cancer~Age + Female + Smokes + Marker1 + Marker2, data = dcaData,thresholds = seq(0, .4, by = .01), bootstraps = 10)
以上代码就完成了两个模型的构建,当然里面的参数可以自行修改。接下来看下模型的构造情况代码:
- summary(baseline.model)
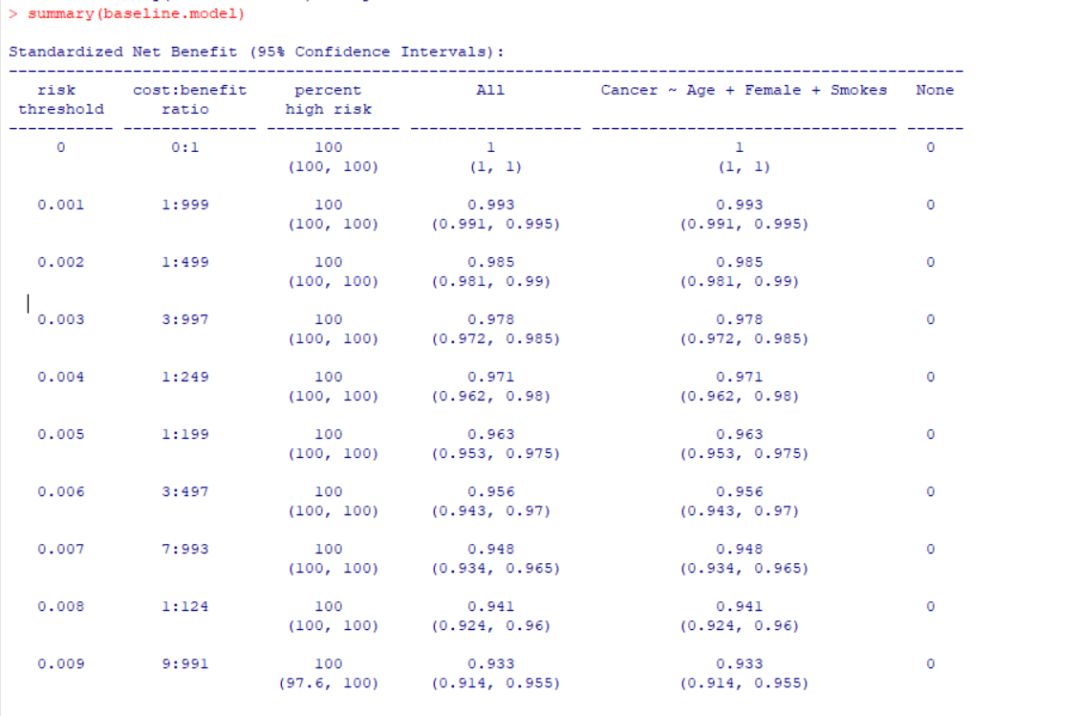
然后就是决策曲线的绘制:
- plot_decision_curve( list(baseline.model, full.model), curve.names = c("Baseline model", "Full model"),col = c("blue", "red"),lty = c(1,2), lwd = c(3,2, 2, 1),legend.position = "bottomright")
结果如图:
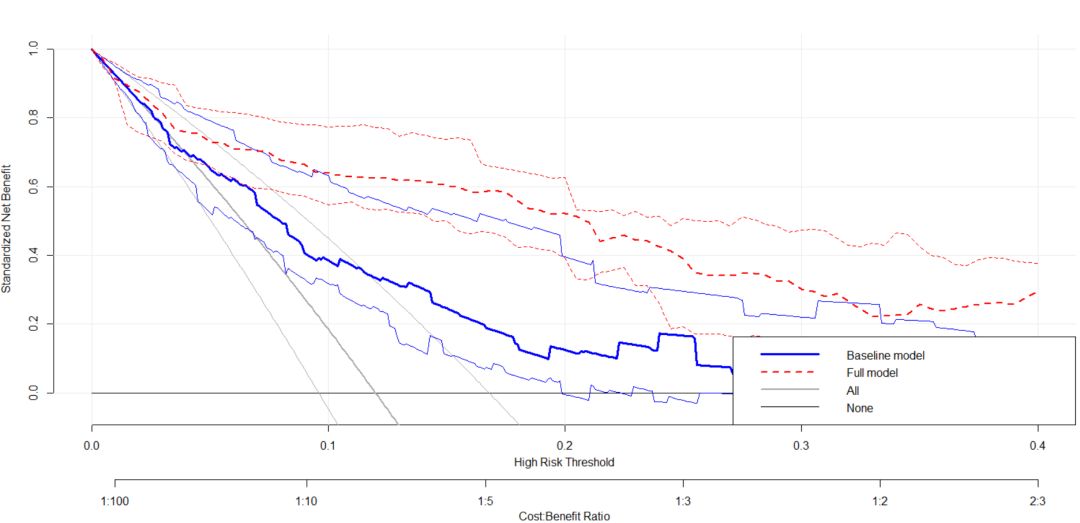
是不是有点乱,那我们把一些辅助线去掉的代码也有:
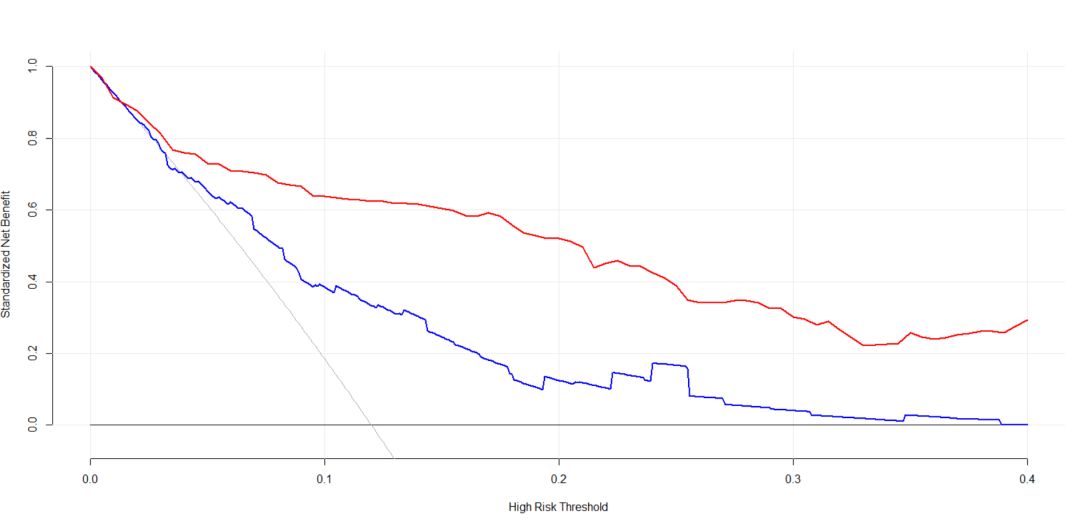
- plot_decision_curve( list(baseline.model, full.model), curve.names = c("Baseline model", "Full model"), col = c("blue", "red"),
- confidence.intervals = FALSE, #remove confidence intervals
- cost.benefit.axis = FALSE, #remove cost benefit axis
- legend.position = "none") #remove the legend
结果如图:
然后是样本数量的评估代码:
- plot_clinical_impact(baseline.model, xlim = c(0, .4), col = c("black", "blue"))
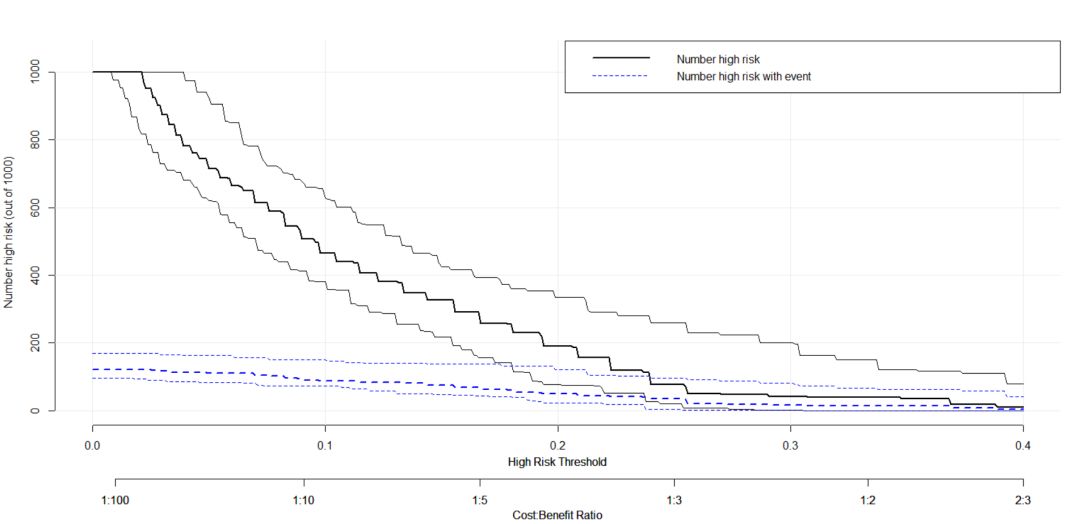
其可以展示高风险样本在模型中生成的情况以及在原始样本中的分布情况,接下来是假阳性评估概率分布:
- plot_roc_components(baseline.model, xlim = c(0, 0.4), col = c("black", "red"))
结果入下:
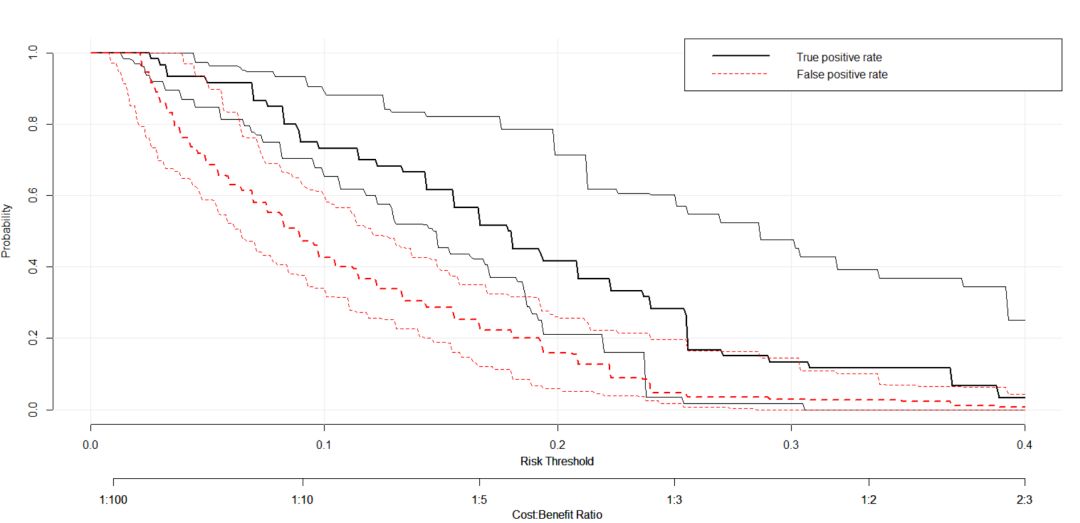
其主函数中包含了利用交叉验证和随机抽样两种方式构建模型的方法,我么也可以看下其两者的差别,此处我们运行的是样例代码,具体的是不是有差别还得看自己的数据。代码如下:
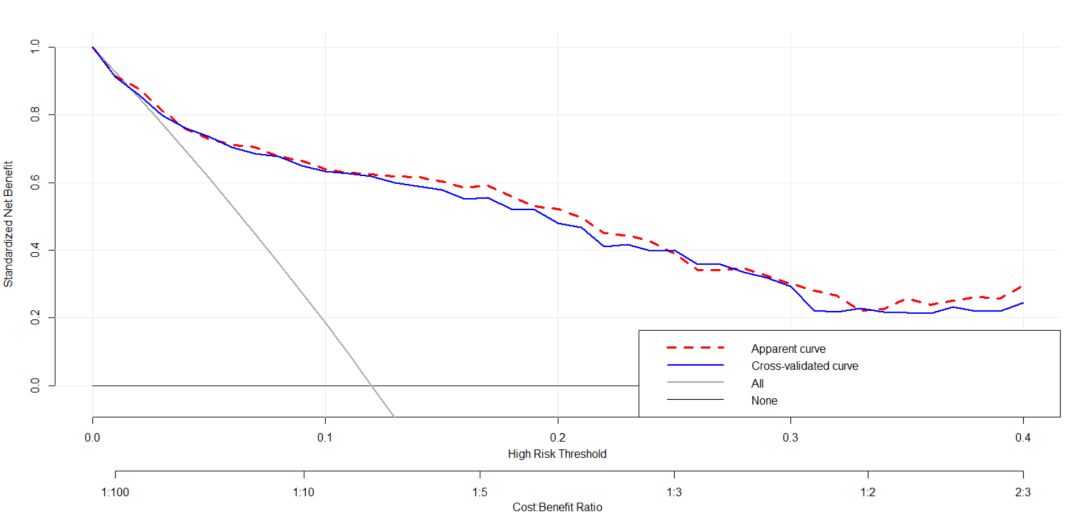
- full.model_cv <- cv_decision_curve(Cancer~Age + Female + Smokes + Marker1 + Marker2, data = dcaData, folds = 5, thresholds = seq(0, .4, by = .01))
- plot_decision_curve( list(full.model_apparent, full.model_cv), curve.names = c('Apparent curve', 'Cross-validated curve'), col = c('red', 'blue'),lty = c(2,1), lwd = c(3,2, 2, 1), legend.position = 'bottomright')
展示结果如下: