

基本概念
WGCNA全名Weighted Gene Co-Expression Network Analysis,粗略翻译为加权基因共表达网络分析,因此可以归于共表达网络分析。
WGCNA主要运用于在大样本基因表达数据,从中挖掘出具有相似表达谱的基因,接着将这些基因聚集在一起,并归于同一模块(module)中。这是由于作者认为具有相同表达趋势的基因,在一些生物学功能上可能也有一定的相关性。然后通过模块特征值(module eigengene)或者hub gene对模块进行区分,接着计算模块与模块之间的相关性以及模块与样本性状之间的相关性,从而筛选有与性状高度相关的模型,并对模块中的基因进行分析,从而找到跟研究相关的目标基因。不少文章运用这方法来寻找潜在生物标志物以及药物靶点。
这种方法跟普通的聚类方法不同点在于其将基因表达值的相关系数取了N次幂,从而使得相关系数分布更加符合无尺度网络分析,这更符合生物学规则。这方法跟一般的共表达网络分析的区别在于其加入了软阈值和权重网络的概念,从而形成加权的共表达网络。
因此WGCNA分析步骤可以分为以下几步:
- 通过数据处理,将基因间的相互作用关系强度符合无尺度分布
- 将基因分类,并把表达模式相似的基因归为一个模块
- 研究模块,找出跟我们研究相关的模块
- 研究模块内基因之间的调控关系
从上四步可以看出,WGCNA应该是:处理大样本(官网建议一般需要至少8个样本以上(不算生物学重复)),筛选出一定量表达模式相同的基因,进而研究基因之间的关系,属于预测范畴。
下面通过一个实例(表达谱文件和样本性状文件)总结下WGCNA的使用流程
分析流程
- 导入表达谱数据和样品性状数据,从表达矩阵可看出总共有3600个基因、136个样本,性状矩阵有361个样本、7个性状
- library(WGCNA)
- options(stringsAsFactors = FALSE)
- dataExpr <- read.csv(file = "Sample_Expr.csv")
- dim(dataExpr)
- [1] 3600 136
- dataTraits <- read.csv(file = "ClinicalTraits.csv")
- dim(dataTraits)
- [1] 361 7
- 数据预处理:我们可以通过过滤低表达量和低变异系数的基因以减少参与后续分析的基因数目,有助于结果的可靠性。由于这个实验数据的特殊性,就不做这步处理了。但我们需要将矩阵变为符合WGCNA要求的形式:行名为gene,列名为样品。并将性状数据和表达谱数据保持一致(一一对应)
- dataExpr <- as.data.frame(t(dataExpr))
- colnames(dataExpr) <- dataExpr[1,]
- dataExpr <- dataExpr[-1,]
- dataExpr <- unlist(apply(dataExpr, 1, as.numeric))
- match_trait <- match(rownames(dataExpr), dataTraits$Mice)
- rownames(dataTraits) <- dataTraits$Mice
- Traits <- dataTraits[match_trait, -c(1,2)]
- 筛选合适的阈值:选一个合适的值,使得样本变为一个无尺度分布,即少部分基因处于绝对优势的位置(表达量高),大部分处于劣势(表达量低)。通过幂指数的方式,对基因之间的相关系数取一定的幂指数,最终确定一个合适的阈值。主要是power值的选择,如下图所示:
- # Choose a set of soft thresholding powers
- powers <- c(c(1:10), seq(from = 12, to=20, by=2))
- sft <- pickSoftThreshold(dataExpr, powerVector = powers)
- # Plot the results:
- par(mfrow = c(1, 2))
- plot(sft$fitIndices[, 1], -sign(sft$fitIndices[, 3]) * sft$fitIndices[, 2],
- xlab = "Soft Threshold (power)", ylab = "SFT, signed R^2", type = "n", main = paste("Scale independence"))
- text(sft$fitIndices[, 1], -sign(sft$fitIndices[, 3]) * sft$fitIndices[, 2],
- labels = powers, col = "red")
- abline(h = 0.9, col = "red")
- plot(sft$fitIndices[, 1], sft$fitIndices[, 5], type = "n", xlab = "Soft Threshold (power)",
- ylab = "Mean Connectivity", main = paste("Mean connectivity"))
- text(sft$fitIndices[, 1], sft$fitIndices[, 5], labels = powers, col = "red")
- 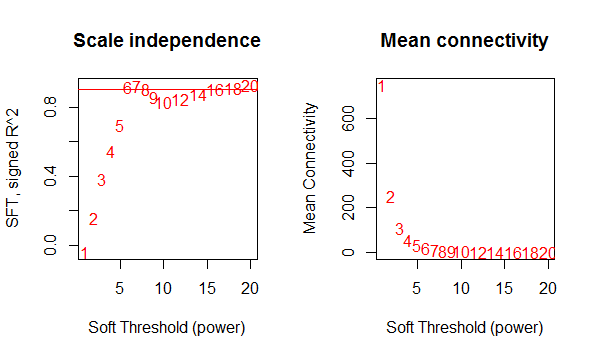
- 根据上图中的红线位置,确定power阈值选为7,即此时的R^2在0.9左右。如果想知道WGCNA建议的阈值,可以输入sft,列表的第一项值就是估计的最优值
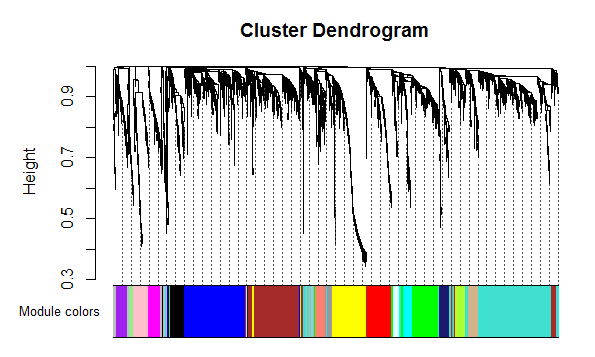
- 通过动态剪切树算法(dynamic tree cutting)分割模块,构建网络图。网给出了两种方法,一个是一步法:简单明了,一步出结果。另一个是分步法:可以细调一系列的参数,以达到满意的结果。官网也没说哪种适合什么条件,都可以用。具体参数设置不讲述了,可以查看这个博客http://blog.sina.com.cn/s/blog_61f013b80101lcpr.html
- net <- blockwiseModules(dataExpr, power = 7, maxBlockSize = 5000,
- TOMType = "unsigned", minModuleSize = 30,
- reassignThreshold = 0, mergeCutHeight = 0.25,
- numericLabels = TRUE, pamRespectsDendro = FALSE,
- saveTOMs = TRUE,
- saveTOMFileBase = "FPKM-TOM",
- verbose = 3)
- moduleLabelsAutomatic <- net$colors
- moduleColorsAutomatic <- labels2colors(moduleLabelsAutomatic)
- # A data frame with module eigengenes can be obtained as follows
- MEsAutomatic <- net$MEs
- 然后展示网络图,如下:
- plotDendroAndColors(net$dendrograms[[1]], moduleColorsAutomatic[net$blockGenes[[1]]],
- "Module colors",
- dendroLabels = FALSE, hang = 0.03,
- addGuide = TRUE, guideHang = 0.05)
- 
- 结合模块与样本性状的相关性来分析,将模块与表型关联
- nGenes <- ncol(dataExpr)
- nSamples <- nrow(dataExpr)
- #same to MEsAutomatic
- MEs0 <- moduleEigengenes(dataExpr, moduleLabelsAutomatic)$eigengenes
- #ME(module eigengene)
- MEs <- orderMEs(MEs0)
- modTraitCor <- cor(MEs, Traits, use = "p")
- modTraitP <- corPvalueStudent(modTraitCor, nSamples)
- 然后做模块与表型相关系数图,如下:
- textMatrix <- paste(signif(modTraitCor, 2), "\n(", signif(modTraitP, 1), ")",sep = "")
- dim(textMatrix) <- dim(modTraitCor)
- par(mar = c(6, 5.5, 3, 3))
- labeledHeatmap(
- Matrix = modTraitCor, xLabels = names(Traits), yLabels = names(MEs),
- ySymbols = names(MEs), colorLabels = FALSE, colors = greenWhiteRed(50),
- textMatrix = textMatrix, setStdMargins = FALSE,
- cex.text = 0.7, zlim = c(-1, 1), main = paste("Module-trait relationships")
- )
- 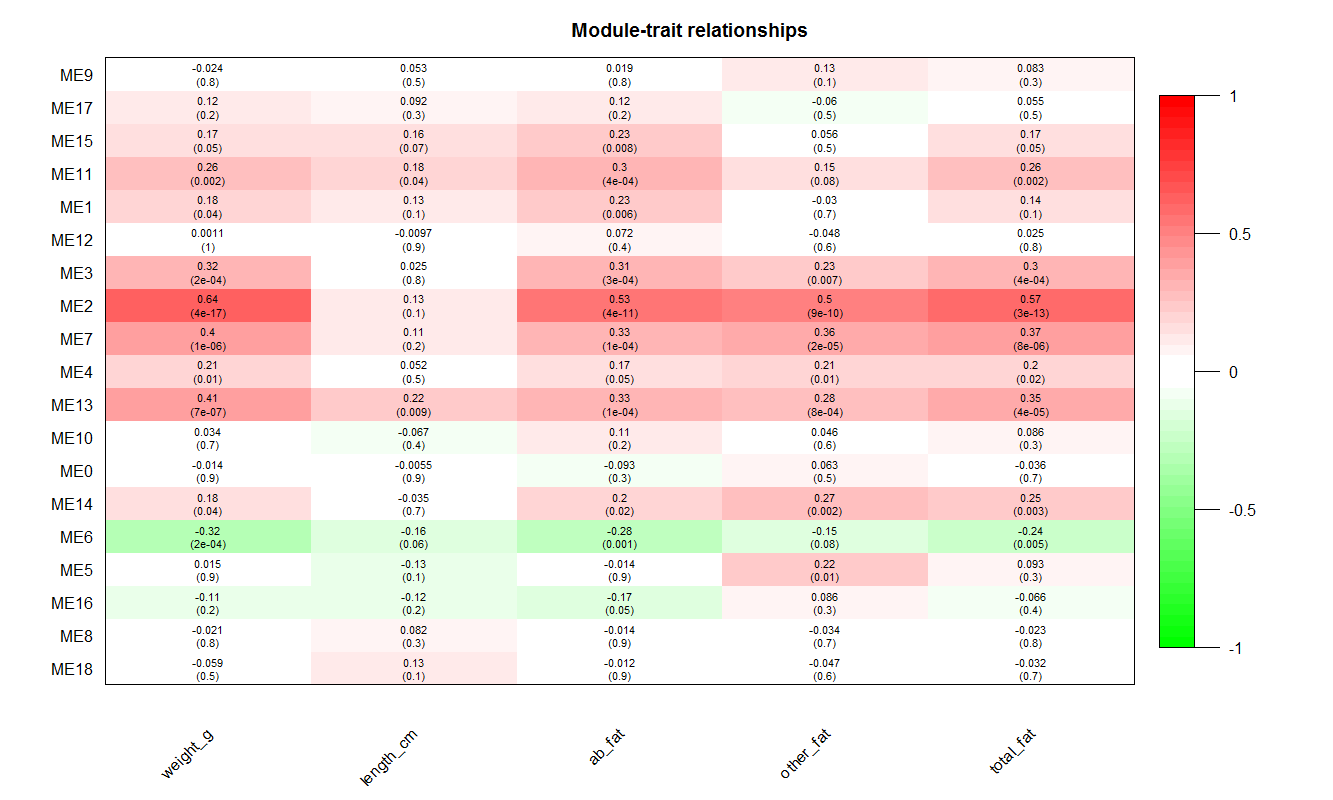
- 图中,y轴是模块名,x轴是性状,每个模块都跟每个性状有一个空格,空格的上部分是模块与性状的相关系数,空格的下部分是一个p值,代表相关系数的显著性。
- 其实随着WGCNA的广泛使用,其用处不仅局限于对性状的分析,还可以结合模块与样本的相关性来分析。比如当研究不同处理组织或者不同发育时期的植物时,如果想了解处理或者不同时期中那些模块对其有显著影响,那么我们可以自行构建类似于性状的Traits矩阵。比如最简单的一种方法就是默认为各个样本间是无关联的,那么Traits矩阵则为标量矩阵,当然还有其他方式(比如考虑样品有生物学重复的情况)。
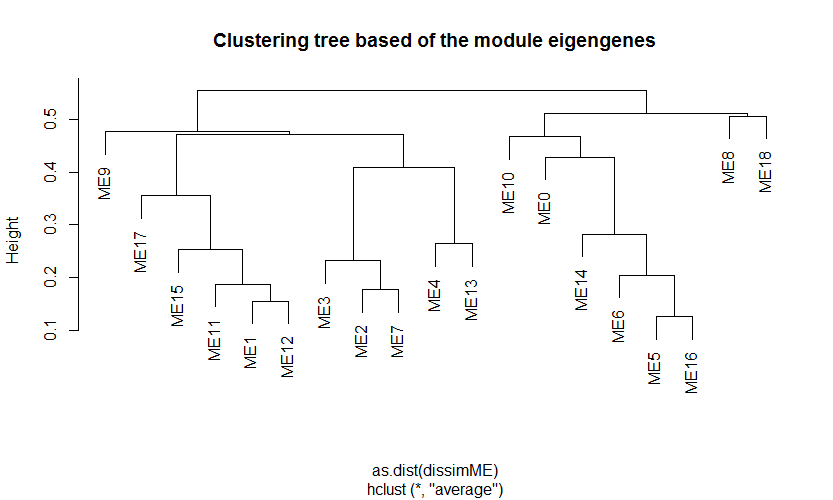
- 查看每个模块之间的相似性以及向异性,并作树杈图,如下:
- dissimME <- (1-t(cor(MEs, method="p")))/2
- hclustdatME <- hclust(as.dist(dissimME), method="average" )
- # Plot the eigengene dendrogram
- plot(hclustdatME, main="Clustering tree based of the module eigengenes")
- 
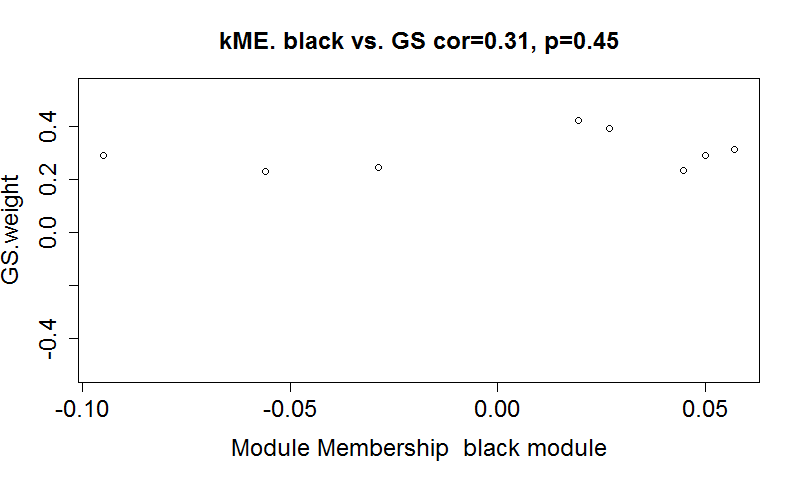
- 查看特定模块中的基因对于特定性状是否具有高GS值(gene significance)和MM值(module membership)。以weigth为例,需要先计算每个基因在每个模块中的KME值,然后再计算GS值,最后作图,如下:
- MEs0 <- moduleEigengenes(dataExpr, moduleColorsAutomatic)$eigengenes
- weight <- as.data.frame(Traits$weight_g)
- names(weight) = "weight"
- GS.weight = as.numeric(cor(dataExpr, weight, use = "p"))
- MEs <- orderMEs(MEs0)
- datKME <- signedKME(dataExpr, MEs)
- ME2Column <- substring(names(MEs), 0)
- column <- match("MEblack", ME2Column)
- restModule = moduleColorsAutomatic == "black"
- verboseScatterplot(MEs[restModule, column], GS.weight[restModule],
- xlab = paste("Module Membership ","black", "module"),
- ylab = "GS.weight", main = paste("kME.", "black", "vs. GS"), col = "black")
- 
- 最后导出模块中TOM值的数据,导入cytoscape作图
- TOM <- TOMsimilarityFromExpr(dataExpr, power = 7)
- # Select modules需要修改,选择需要导出的模块颜色
- modules <- c("black")
- # Select module probes
- probes <- colnames(dataExpr)
- inModule <- is.finite(match(moduleColorsAutomatic, modules))
- modProbes <- probes[inModule]
- # Select the corresponding Topological Overlap
- modTOM <- TOM[inModule, inModule]
- dimnames(modTOM) <- list(modProbes, modProbes)
- # Export the network into edge and node list files Cytoscape can read
- cytoscape <- exportNetworkToCytoscape(modTOM,
- edgeFile = paste("FPKM-One-step-CytoscapeInput-edges-", paste(modules, collapse="-"), ".txt", sep=""),
- nodeFile = paste("FPKM-One-step-CytoscapeInput-nodes-", paste(modules, collapse="-"), ".txt", sep=""),
- weighted = TRUE,
- threshold = 0.2,
- nodeNames = modProbes,
- nodeAttr = moduleColors[inModule])
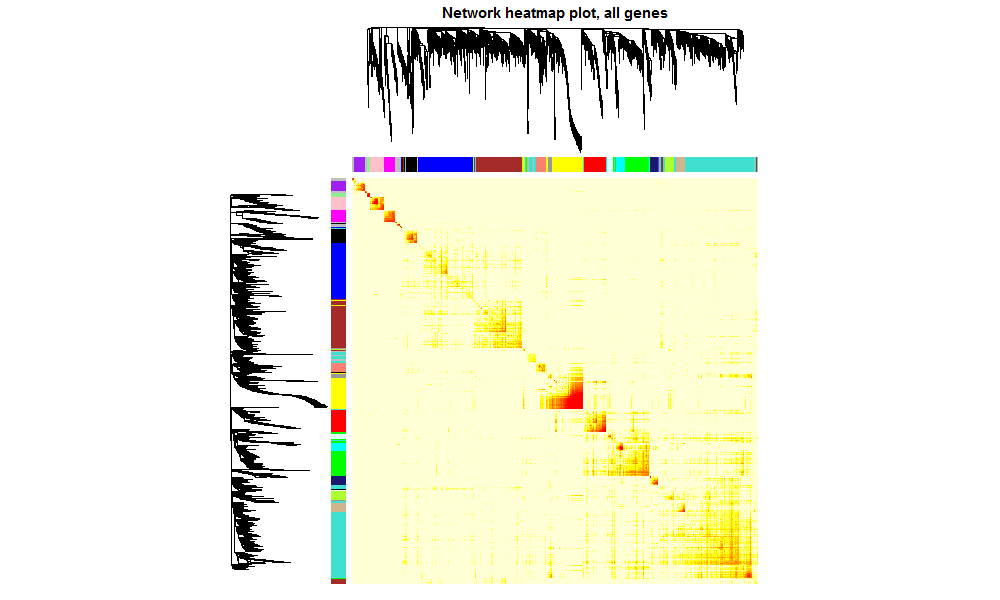
- 并做一个TOM图,如下:
- dissTOM <- 1 - TOM
- plotTOM = dissTOM^7
- diag(plotTOM) <- NA
- geneTree <- net$dendrograms[[1]]
- TOMplot(plotTOM, geneTree, moduleColorsAutomatic, main = "Network heatmap plot, all genes")
- 
- 最后就是用cytoscape将上述特定模块中的基因以互作网络图形式展现,以一定的标准来选择hub gene作为后续研究的重点也可以对模块中的每个基因进行注释,做GO以及KEGG富集。根据富集到的通路,结合模块信息来确定哪个模块的基因作为后续研究的重点