

这篇文章最好的地方不只在于推荐了工具,提供了一套分析流程,更在于详细介绍了有哪些方法可用、什么时候用哪些方法和有什么注意事项。这也是我们易生信培训过程中广泛讨论的问题。
Abstract
单细胞RNA-seq使研究者能够以前所未有的分辨率研究基因表达图谱。这一潜力吸引着更多科研工作者应用单细胞分析技术解决研究问题。随着可用的分析工具越来越多,如何组合成一个最新最好的数据分析流程也越来越难。我们详细阐述了一个典型的单细胞转录组分析各个步骤的细节和注意事项,包括预处理(质控、标准化、数据校正、特征选择、降维)和细胞/基因水平的下游分析等。基于独立的比较研究,我们为每一步都推荐了当前最好的方法和操作建议。随后把这些最好的工具整合成一个分析流程并应用于一套公共数据集的分析以演示其效果。案例研究具体可见https://www.github.com/theislab/single-cell-tutorial。这篇综述为这个领域的新人提供了一份学习单细胞分析的指南,并且也能帮助老用户更新他们的分析流程。
背景
近年来,单细胞RNA测序(scRNA-seq)大大提高了我们对生物系统的了解。我们已经能够在研究斑马鱼、青蛙和涡虫(zebrafish, frogs and planaria)等生物细胞异质性的同时发现先前未知的细胞群体。这项技术的巨大潜力激励了计算生物学家开发了一系列分析工具。尽管开发者为了确保单个工具的可用性付出了巨大的努力,但是由于该领域的相对不成熟,对于单细胞数据分析的新手来说,入门的障碍是缺少一份标准指南。在本文中,我们提供了一份scRNA-seq分析的参考教程,并概述了当前的最佳实践方案,为将来的分析标准化奠定了基础。
分析标准化面临的挑战来源于越来越多的可用分析方法(截至2019年3月7日有385种工具)和数据集规模爆炸性的提高。使得我们一直在寻找新的方法来分析处理我们的数据。例如,最近已经有方法可以预测细胞分化过程中的命运选择。尽管分析工具的不断改进有助于产生新的科学推论,但它也使分析流程的标准化变得更为复杂。
标准化的另一个挑战在于软件技术方面。用于scRNA-seq数据的分析工具是用不同的编程语言编写的 - 最主要的是R和Python。尽管跨编程语言的支持越来越多,但使用的编程语言确实影响了对分析工具的选择。诸如Seurat,Scater或Scanpy等常用工具提供了集成环境来开发流程并包含大量分析工具。然而,出于维护的需要, 这些平台限制了它们只能使用各自的编程语言开发的工具。引申开来,当前可用的scRNA-seq分析教程也存在语言限制,而且许多是围绕这3大平台进行讲述的 (易生信2020年最新的单细胞课程会同时提供R和Python版本的最新分析流程):
- R and bioconductor tools:
https://github.com/drisso/bioc2016singlecell;
https://hemberg-lab.github.io/scRNA.seq.course/; https://www.ncbi.nlm.nih.gov/pubmed/27909575?dopt=Abstract; - Seurat: https://satijalab.org/seurat/get_started.html;
- Scanpy: https://scanpy.readthedocs.io/en/stable/tutorials.html;
考虑到上述挑战,我们选择概述当前分析过程中每一步的最佳实践和独立于编程语言的常用工具,而不是评估一个完整的分析流程。我们将带领读者完成scRNA-seq分析流程的各个步骤,介绍每个步骤的最优方法,并讨论分析过程中会遇到的陷阱和当前未解决的问题。当然由于一些工具比较新,并且缺乏系统比较,最好的工具很难决定,我们列出了流行的可用工具。列出的工具从基因count矩阵开始到潜在的分析终点。在我们的Github上有结合了已建立的当前最佳实践流程的案例研究:https://github.com/theislab/single-cell-tutorial/。我们在实际的示例工作流程中应用了当前的最佳实践来分析公共数据集。该分析流程使用Jupyter notebook和rpy2整合了R和Python的工具。结合现有的文档,这已经可以被视作一个可接受的分析流程模板了。
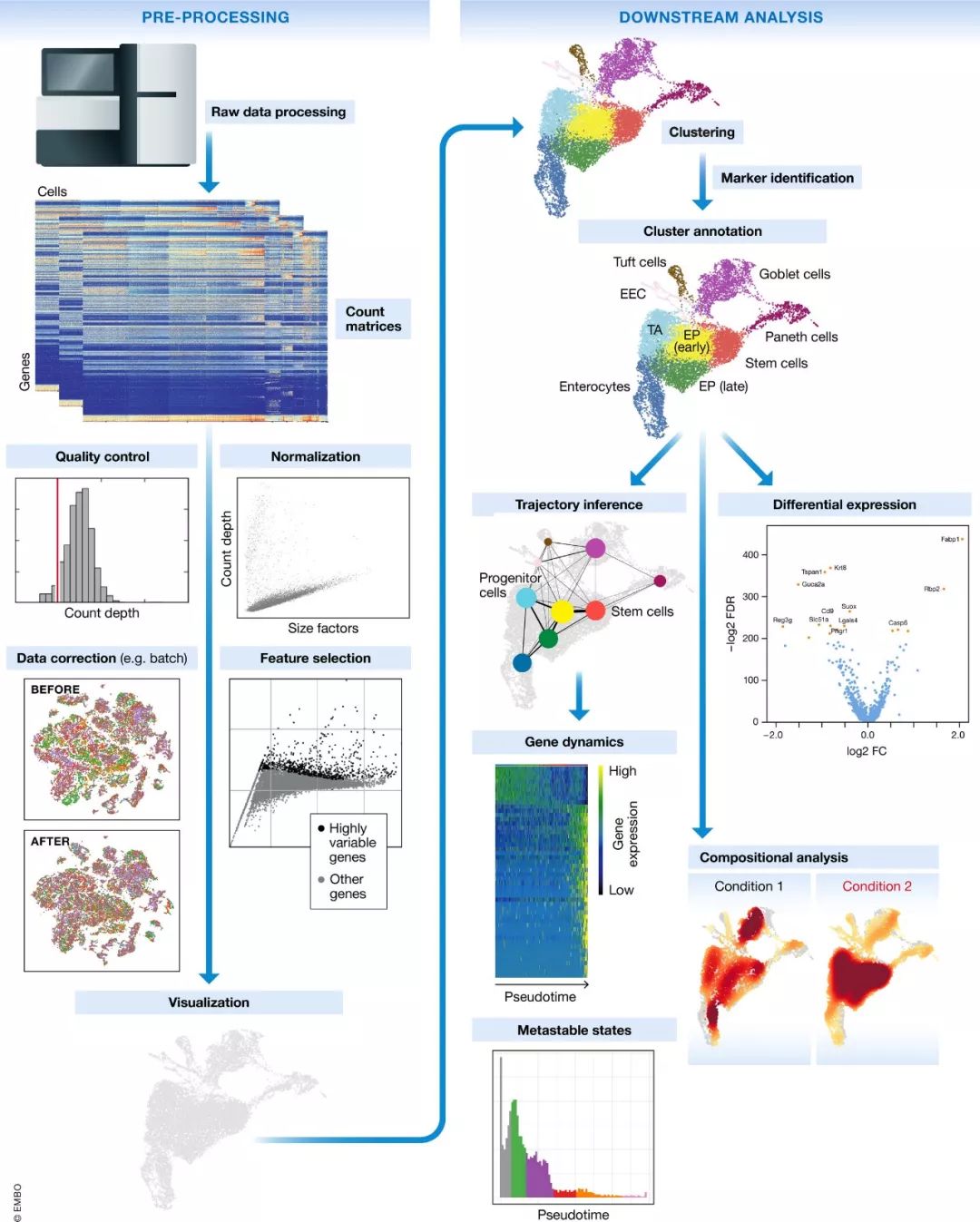
图1. 典型单细胞转录组分析流程。
原始测序数据预处理和比对后获得Gene count矩阵,作为本分析流程的开始。这些结果图是Count矩阵采用最佳实战分析流程进行预处理和下游分析获得的。数据来源于Haber et al 2017肠上皮细胞文章。
预处理和可视化
原始测序数据经过处理得到分子计数矩阵(count matrix),或者reads count (读数矩阵)。这取决于单细胞文库构建方案中是否包含唯一分子标识符(UMI, unique molecular identifiers)(参考下面的Box1. Hemberg-lab单细胞转录组数据分析(六)- 构建表达矩阵,UMI介绍)。原始数据处理流程如Cell Ranger , indrops, SEQC, zUMIs负责测序数据质控,确定reads来源的细胞 (barcodes) (这一步也称为demultiplexing)和mRNA分子 (生信宝典注:单细胞测序不只获得mRNA,更准确说是带poly-A尾巴的RNA,包括mRNA和lncRNA等)、基因组比对和定量。获得的reads或count矩阵的行数等于barcodes的数目,列数等于基因数目(生信宝典注:也可能反过来,行为基因,列为barcodes)。这里使用术语barcodes而不是cell,因为分配给相同barcode的所有reads可能并不只是来源于同一细胞。barcode可能会错误地标记多个细胞(doublet),也可能不会标记任何细胞(空液滴/孔)。
尽管reads data和 count data 的测量噪声级别不同 (生信宝典注:基于UMI的数据,获得的是分子计数,count data,噪声更低),但在分析流程中的处理步骤是相同的。为简单起见,在本教程中,我们将数据称为计数矩阵(count data)。在reads和计数矩阵的结果不同的地方,将会特别提到reads矩阵。
Box1:scRNA-seq实验流程的关键要素
从生物样品到单细胞数据需要多步实验操作。典型的工作流程包含单细胞解离、单细胞分离、文库构建和测序。我们在这里简要介绍这些步骤。不同protocols的更详细的解释和比较可参考下面三篇文章:
Ziegenhain et al (2017):https://www.embopress.org/doi/10.15252/msb.20188746#msb188746-bib-0159
Macosko et al (2015):https://www.embopress.org/doi/10.15252/msb.20188746#msb188746-bib-0083
Svensson et al (2017):https://www.embopress.org/doi/10.15252/msb.20188746#msb188746-bib-0126
- 单细胞实验的输入材料通常是生物组织样品。第一步,单细胞解离:消化组织产生单细胞悬液。为了分别分析每个细胞中的
mRNA,必须分离单细胞。根据实验方案不同,单细胞分离的方式也有所不同。
- 基于平板的技术将细胞分到到板上的孔中。
- 基于液滴的方法则依赖于微流体液滴捕获单个细胞。
- 在这两种情况下,都可能出现一些问题,如多个细胞一起被捕获(
doublets or multiplets)、非活细胞被捕获或根本没有细胞被捕获(空液滴/孔)。基于液滴的方法需要通过低的输入细胞浓度来保持低的doublets率,因此空液滴是特别常见的 (生信宝典注:一般beads和细胞的输入比例是20:1)。
每个孔或液滴均包含必要的试剂以裂解细胞膜并进行文库构建 (生信宝典注:植物单细胞就要注意了,需要提前去除细胞壁)。文库构建包括捕获细胞内mRNA、反转录为cDNA分子并进行扩增等过程。因为文库构建时每个细胞是独立的,所以每个细胞的mRNA也就特异的标记了孔特异性或液滴特异性细胞barcode。此外,许多实验方案还使用唯一分子标识符(UMI)标记捕获的RNA分子。一般在测序之前需要先扩增细胞cDNA以增加其被检测的可能性。但微量扩增更容易引入PCR偏好性。UMI使我们能够区分测到的reads是来源于mRNA分子的不同扩增拷贝还是来源于独立的mRNA分子,从而可以进行更准确的定量。
每个细胞单独构建的cDNA文库都带有cell barcode和/或UMI(取决于protocol),后续将这些文库混合在一起测序。测序产生的reads数据进行质量控制,根据其barcodes序列分组(demultiplexing),并且进行后续比对定量。对基于UMI的protocols,reads的数据可以进一步demultiplexed以得到捕获的mRNA分子的计数(count data)。也就是本套流程的起始输入数据。
质控
在分析单细胞基因表达数据之前,我们必须确保所有barcode都对应于有效细胞 (viable cells,有活力的细胞)。质控有3个指标:
- 测到的转录本分子总数
- 测到的基因总数
- 来源于线粒体基因的转录本所占比例
质控就是检查这3个指标的分布中是否存在异常峰并设置阈值去除。这些异常的barcodes可能对应于死细胞、细胞膜破损的细胞或doublets。比如,如果某个barcode对应的样品测到的分子总数低、检测到的基因数少、线粒体基因所占比例高,则表明该样品可能存在细胞膜破损导致细胞质RNA漏出,只有线粒体中的RNA保留了下来。相反,如果某个barcode对应的样品有异常高的总分子数和检测到的基因数,则有可能这个样品包含2个或以上细胞(doublets)。因此,高的总分子数通常用来过滤潜在的doublets。3个最近发表的doublets检测工具提供了更好的解决方案:DoubletDecon, Scrublet, Doublet Finder。
分开考虑这三个QC变量中的任何一个都可能导致对细胞状态的错误解读。例如,线粒体计数相对较高的细胞可能是呼吸过程比较活跃 (生信宝典注:如心脏细胞总mRNA的30%是线粒体基因,具体见对单细胞RNA综述的评述:细胞和基因质控参数的选择)。同样,其他QC变量也具有相应的生物学意义。总分子数和/或基因数低的细胞可能是处于静息状态的细胞群体;总分子数和/或基因数高的细胞也可能是细胞自身体积较大。实际上,细胞之间的总分子数自身也可能有很大差异(具体见Github上的案例研究)。因此,在做出是否过滤的单阈值决策时,应联合考虑3个QC变量,并且应将这些阈值设置的尽可能宽松,以避免无意间滤除有效的细胞群。将来,考虑多元QC依赖的过滤模型可能会提供更敏感的QC选择方式。
包含不同类型细胞混合体的数据集的每个QC指标可能会呈现多个分布聚集峰。例如,图2D显示了具有不同QC分布的两个细胞群。如果未执行先前的过滤步骤(请注意,Cell Ranger也会执行细胞QC),则应仅将最低总分子数和基因数的barcode视为无效细胞。另一个阈值设定准则是考虑所选阈值过滤掉的细胞比例。比如过滤高分子总数细胞时,过滤掉的细胞比例不应超过预期的doublet率。
除了检查细胞的完整性外,还必须在转录本水平上执行QC步骤。原始计数矩阵通常包含超过20,000个基因。可以通过滤除在多数细胞中不表达的基因 (通常认为这些基因对细胞异质性分析贡献不大),大大减少该数目。设置此阈值的准则是使用感兴趣的最小细胞亚群大小,并为dropout事件留出一些余地 (生信宝典注:比最小细胞亚群大小数字再小一点)。例如,滤除掉在少于20个细胞中表达的基因可能会使鉴定少于20个细胞的细胞亚群变得困难。对于dropout比率高的数据集,此阈值也可能会使不太大的细胞亚群难以被检测到。阈值的选择应随待分析数据集中的细胞数量和既定的下游分析而定。
另外可以直接对计数矩阵 (count matrix)执行进一步的质量控制。输入型基因表达(Ambient gene expression)是指某个barcode检测到的转录本是源自其他细胞在建库前发生破裂而释放到细胞悬液中的mRNA。这些增加的外来计数会影响下游分析,如Marker基因鉴定或其他差异表达分析过程。由于基于液滴的scRNA-seq数据集中存在大量空液滴,因此可以通过空液滴建模分析细胞悬液中的RNA构成和丰度来校正这一影响。最近开发的SoupX使用这种方法直接校正count matrix。另外,在下游分析中直接忽略这些有强影响的输入型基因也是处理这个问题的一个实用方法。
质控是用于保证下游分析时数据质量足够好。由于不能先验地确定什么是足够好的数据质量,所以只能基于下游分析结果(例如,细胞簇注释)来对其进行判断。因此,在分析数据时可能需要反复调整参数进行质量控制 (生信宝典注:反复分析,多次分析,是做生信的基本要求。这也是为啥需要上易生信培训班而不是单纯依赖公司的分析。)。从宽松的QC阈值开始并研究这些阈值的影响,然后再执行更严格的QC,通常是有益的。这种方法对于细胞种类混杂的数据集特别有用,在这些数据集中,特定细胞类型或特定细胞状态可能会被误解为低质量的异常细胞。在低质量数据集中,可能需要严格的质量控制阈值。数据集的质量可以通过实验质量控制指标来确定 (see Appendix Supplementary Text S2,简单来说就是原始数据测序质量(FastQC的结果、序列比对率和比对到已经注释的基因区的reads比率、实际检测到的细胞数和预估细胞数是否吻合)。在QC阈值迭代优化过程中,要避免数据挑选 (data peeking)。QC阈值不应用于改善统计检验的结果。相反,可以根据数据集可视化和聚类中QC变量的分布来评估QC选取的阈值是否合理。
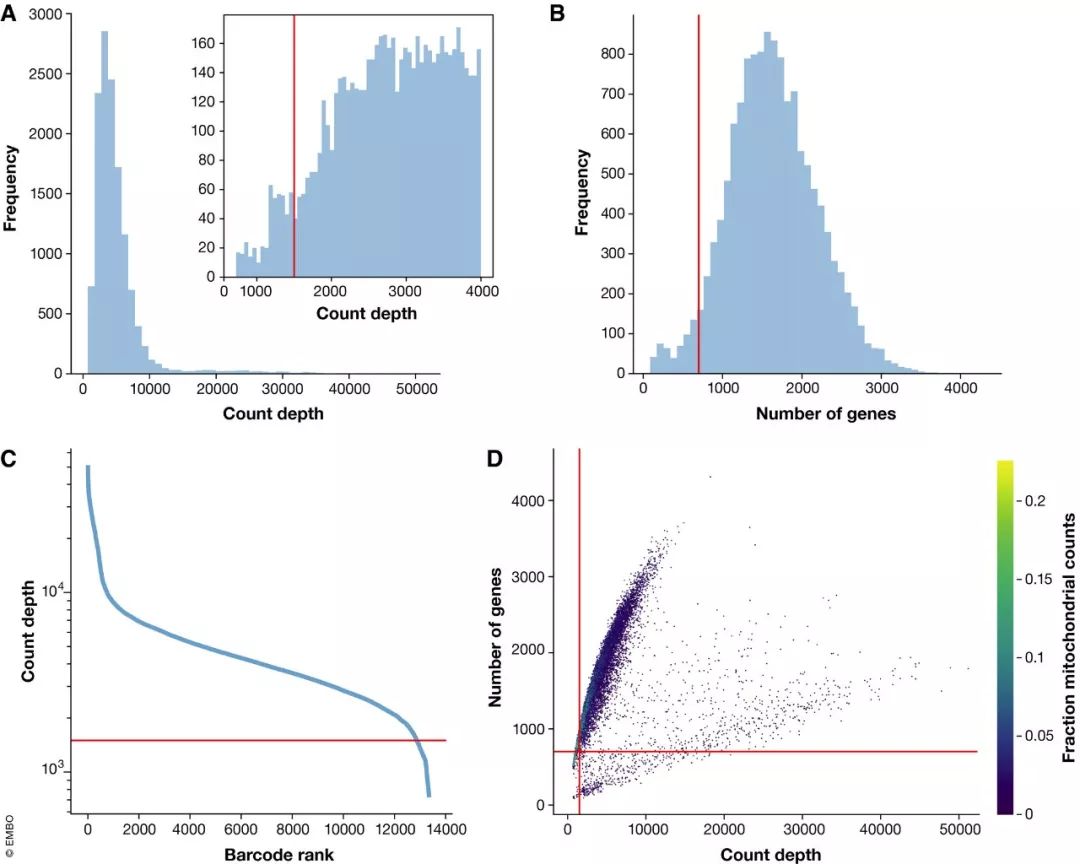
图2. 质控变量的分布图 (小鼠肠道上皮细胞数据集)
(A) 每个细胞检测到的总分子数的分布用直方图展示。右上角的子图展示了总分子数小于4,000的部分的分布。因为在count数为1200处有个峰,所以阈值设置为了1,500。(B) 每个细胞检测到的基因数的直方图展示。在400个基因处有个小峰(噪音峰),阈值设置为700。(生信宝典注:这阈值设置的随意性也没谁了~~~)(C) 每个细胞检测到的总分子数从高到底绘制rank plot,类似于Cell Ranger检测并过滤空液滴的log-log plot。在总分子数为1500时,存在一个快速降低的拐点,则1500为筛选阈值。(D) 同时展示检测到的基因数(纵轴)、总分子数(横轴)和线粒体基因的比例 (颜色)。线粒体基因只在低基因数和低总分子数的细胞中比例高。这些细胞已经被上面设置的总分子数和基因数阈值过滤掉了。质控参数联合可视化显示更低的基因数筛选阈值可能也是足够的。
陷阱和建议:
- 通过可视化检测到的基因数量、总分子数和线粒体基因的表达比例的分布中的异常峰来执行QC。联合考虑这3个变量,而不是单独考虑一个变量。
- 尽可能设置宽松的QC阈值;如果下游聚类无法解释时再重新设定严格的QC阈值。
- 如果样品之间的QC变量分布不同(存在多个强峰),则需要考虑样品质量差异,应按照
Plasschaert et al. (2018)的方法为每个样品分别确定QC阈值。
标准化
计数矩阵中的每个数值代表细胞中一个mRNA分子被成功捕获、逆转录和测序(Box 1)。由于每个操作步骤固有的可变性,即便同一个细胞测序两次获得的计数深度也可能会有所不同。因此,当基于原始计数数据比较细胞之间的基因表达时,得到的差异可能来自于技术原因。Normalization可以通过调整计数数据 (scaling count data)等解决这一问题,以获得细胞之间可比的相对基因表达丰度。
普通转录组分析有很多可用的标准化方法。尽管其中一些方法已应用于scRNA-seq分析,但单细胞数据特有的变异特征(例如technical dropouts: zero counts due to sampling )催生了scRNA-seq特异性标准化方法的发展。
最常用的标准化方法是测序深度标准化,也称为“每百万计数”或CPM normalization。该方法来自普通转录组表达分析,使用每个细胞的测序深度作为size factor对计数数据进行标准化。CPM标准化假设数据集中的所有细胞最初都包含相等数量的mRNA分子,并且计数深度差异来源于技术问题。这一方法的变体有:把size factor放大10^6倍如CPM,或size factor乘以数据集中所有细胞的测序深度的中位数。downsampling方法也基于同样的假设,从原始数据中随机抽取预设的等量reads以保证所有细胞测序深度相同。downsampling方法在扔掉一些数据的同时增加了technical dropout rates,而CPM或其它全局的标准化方式则不会影响。因此downsampling方法可能更真实地反应了相同测序深度下不同细胞的表达谱特征。
由于单细胞数据集通常由大小和分子数不同的异质细胞群体组成,因此通常需要更复杂的标准化方法。例如,Weinreb et al对CPM算法进行了扩展,在计算size factors时排除在任何细胞中总计数大于5%的基因。这一方法屏蔽掉少数高表达基因对总体表达变化的影响。软件包Scran的pooling‐based size factor方法对细胞异质性的影响处理更好。首先把细胞合并到一起避免technical dropout效应,然后基于基因表达的线性回归模型估算size factor。这一方法允许细胞有少于50%的差异表达基因,并且在不同的测试评估研究中这一标准化方法都表现最好。评估发现,scran比其他标准化方法对后续批次校正和差异表达分析效果更好。并且在scran作者小范围的比较中也展现出这个方法稳定性比较好。
CPM, high‐count filtering CPM和scran 使用线性全局缩放对计数数据进行标准化。还有非线性标准化方式可以处理更多的混杂因素的影响。许多此类方法涉及对计数数据进行参数化建模。例如, Mayer et al 使用技术相关协变量(例如测序深度和每个基因的计数)作为模型参数对count data拟合负二项模型。模型的残差作为标准化后的基因表达值。这种方法可以在标准化的同时校正技术差异和生物来源的差异(例如批次校正或对细胞周期影响的校正)。已表明,非线性标准化方法优于全局标准化方法,尤其是在有较强批次效应影响的情况下。因此,非线性标准化方法特别适用于基于板的scRNA-seq数据,因为不同板之间往往会产生批次效应。此外,基于板的数据与基于液滴的数据相比,每个细胞的测序深度变化更大。理论上非线性标准化方法或诸如downsampling的放法更适合于板的数据 (plate),但仍需要进行比较研究以确认这一观点。在本教程中,我们倾向于将标准化和数据校正(批处理校正、噪声校正等)步骤分开,以强调数据的不同处理阶段。因此,我们专注于全局缩放标准化方法。
我们不能期望一个标准化方法适用于所有类型的scRNA-seq数据。例如,Vieth et al 表明,reads data和count data适用不同的模型。确实,Cole et al 发现不同的标准化方法只对适合的数据集表现最佳,并认为应使用他们的scone工具对数据集进行评估以选择适用的标准化方法。此外,scRNA-seq技术可分为全长和3'富集方法。全长方案的数据可能会受益于考虑了基因长度的标准化方法,而3'富集测序数据则不然。TPM归一化是全长scRNA-seq数据常用的归一化方法,它来自bulk RNA-seq分析。
标准化是对细胞计数数据进行缩放处理以使其在细胞之间可比,也可以在基因层面对基因计数进行归一化 (scale)以便于基因内部进行直接比较。基因归一化是指一个基因减去其在所有样品表达的均值然后除以其在所有样品表达值的标准差。归一化后,这个基因在所有样品表达值均值为0,用单位方差形式表示其表达值。归一化后,所有基因在下游分析时权重是一样的。(生信宝典注:归一化后,原始基因的表达丰度信息就没了,换成了无标度的标准差的表示。)是否对基因进行归一化目前尚无达成共识。尽管流行Seurat教程通常应用gene scaling,但Slingshot方法的作者在其教程中选择了不对基因进行scaling。两种选择的争议点在:所有基因不论表达高低在进行下游分析时权重一致 (生信宝典注:scale后所有基因权重一致),还是基因表达量的绝对值对下游分析也有贡献 (生信宝典注:突出高表达基因对下游的更大影响) 。为了从数据中保留尽可能多的生物相关信息,在本教程中,我们选择不进行归一化 (scaling genes)这一步操作。
标准化后,数据矩阵通常进行log(x+1)转换。此转换具有三个重要作用。
- 首先,对数转换后的表达式值之间的差值可对应于对数转换后的倍数变化,这是衡量基因表达变化的常用方法。
- 其次,对数转换可减轻(但不能消除)单细胞数据的均值-方差关系 (
mean-variance relationship) (生信宝典注:均值-方差关系展示数据在特征空间的分布关系。方差越大数据分布越广,后续采用线性回归算法时效果越差。)。 - 最后,对数转换可以减少数据的偏态分布,从而使数据近似于正态分布,更符合许多下游分析工具对数据分布的假设要求。
尽管scRNA-seq数据实际上不是对数正态分布的,但这三个效果使对数转换成为一种粗略但有用的工具。这一有用性在下游差异表达分析或批次校正时有更好的体现。但是应该注意的是,数据的对数转换会在数据中引入虚假的差异表达结果。而且如果size factor在组间差异更大时影响尤其明显。
陷阱和建议:
- 我们建议使用
scran来标准化非全长数据集。另一种选择是通过scone评估标准化方法,尤其是对基于板的数据集。全长scRNA-seq数据可以使用bulk转录组的方法校正基因长度的影响。- 基因表达归一化(
scale)到均值为0和单位方差尚无共识。我们倾向于不进行scale操作。- 标准化后的数据应进行
log(x+1)转换,以使数据更符合正态分布,满足下游分析方法的初始假设。
数据校正和整合
如前所述,数据标准化是去除实验过程中随机性的影响 (count sampling)。但是,标准化后的数据可能仍然包含有与研究不相干的因素带来的影响。数据校正的目的就是进一步去除技术因素和非关注的生物学混杂因素,例如批次、dropout或细胞周期不同带来的影响。需要注意的是这些混淆因素并不总是需要校正。相反,考虑在分析中移除哪些因素取决于下游分析目的。我们建议分别考虑对生物学和技术混杂因素 (covariates)的校正,因为它们用于不同的目的并代表不同的分析挑战。
去除与研究不相干的生物因素的影响
尽管校正实验中技术因素的影响可能对揭示潜在的生物学信号至关重要,但对不关注的生物学因素的校正可用来挑选出特定的感兴趣的目标生物学信息。最常见的生物因素校正是消除细胞周期对转录组中基因表达的影响。可以使用Scanpy和Seurat对每个细胞的细胞周期评分进行简单的线性回归校正或通过应用了更复杂的混合模型的专用程序包如scLVM或f-scLVM进行校正。用于计算细胞周期评分的标记基因列表可在文献中获取。这些方法还可用于校正其他已知的生物因素的影响,例如线粒体基因表达,通常是细胞应激的标识。
在校正特定的生物因素影响之前,应考虑几个方面。首先,校正生物学因素影响并不总是有助于解释scRNA-seq数据。虽然消除细胞周期影响可以改善对发育轨迹的推断,但细胞周期信号也可以提供有意义的生物学信息。例如,可以根据细胞周期评分确定增殖细胞群 (在Github的案例研究中有讲)。同样,必须根据具体研究的对象理解生物学信号。假设生物过程发生在同一有机体内,则这些过程之间可能存在依赖性。因此,校正一个过程的影响可能会无意间掩盖另一个过程的信号。另外,也有研究者提出细胞大小对转录组的影响通常也归因于细胞周期不同。因此,通过标准化或专用工具(例如cgCorrect)在校正细胞大小的同时也部分校正了scRNA-seq数据中细胞周期的影响。
去除技术影响
用于移除不相干生物因素影响的回归模型也可应用于校正实验技术因素的影响。单细胞数据中最突出的技术影响因素是测序深度和批次。尽管标准化可以使得细胞之间的基因定量数据可比,但测序深度的影响仍保留在数据中。这种测序深度效应既可以是生物学自身的影响,也可能是技术带来的影响。例如,细胞大小可能不同,因此mRNA分子数量也可能不同。而且,标准化后技术带来的计数影响可能依然存在,因为没有办法推断由于实验过程中的随机性带来的未检测到的基因的表达。对测序深度进行回归校正可以提高轨迹推断算法的性能,该算法依赖于查找细胞之间的过渡(具体见Github上的案例研究)。在校正多个协变量(干扰因素)(例如细胞周期和测序深度)时,应在单个步骤中对所有协变量(干扰因素)执行回归,以解决协变量(干扰因素)之间的依赖性。
消除测序深度影响的另一个策略是使用更严格的标准化程序,例如downsampling或非线性归一化方法 (见前面标准化部分)。这些方法可能更适用于基于板的scRNA-seq数据集,因为细胞之间较大的测序深度差异可能掩盖细胞之间的异质性。
批次效应和数据整合
当将细胞分组操作时可能会带来批次效应,比如不同芯片上的细胞、不同测序通道中的细胞或在不同时间点收集的细胞都归类于不同的组。实验操作过程中细胞所经历的不同环境可能会影响转录组的测量结果或甚至影响细胞自身的转录变化。所产生的影响存在多个层面:同一实验不同的细胞组、同一实验室的不同实验或不同实验室的数据集之间。在这里,我们把第一种情况与后面两种情况区分开。校正同一实验中样品或细胞之间的批次效应是bulk RNA测序批次效应的一种经典方案。我们将其与整合来自多个实验的数据(称为数据整合)区分开。通常批次效应校正使用线性方法,而非线性方法则用于数据整合。
最近对经典批次校正方法的比较显示,ComBat在中低到中复杂度的单细胞实验中也表现良好。ComBat构建了基因表达的线性模型,其中批次贡献在数据的均值和方差中均得到校正(图3)。与采用什么计算方法无关,批次校正的最佳方法是通过巧妙的实验设计来避免存在不同批次。通过合并不同实验条件和样品的细胞一起进行后续操作可以避免批次效应。使用细胞标记等策略或根据基因组变异信息,可以对实验中合并的细胞进行拆分。(生信宝典注:最不建议的实验设计方式是对照组样品是一个批次,处理组样品是一个批次;如果这么做,将不能确定差异是来源于批次,还是真的存在生物差异。)
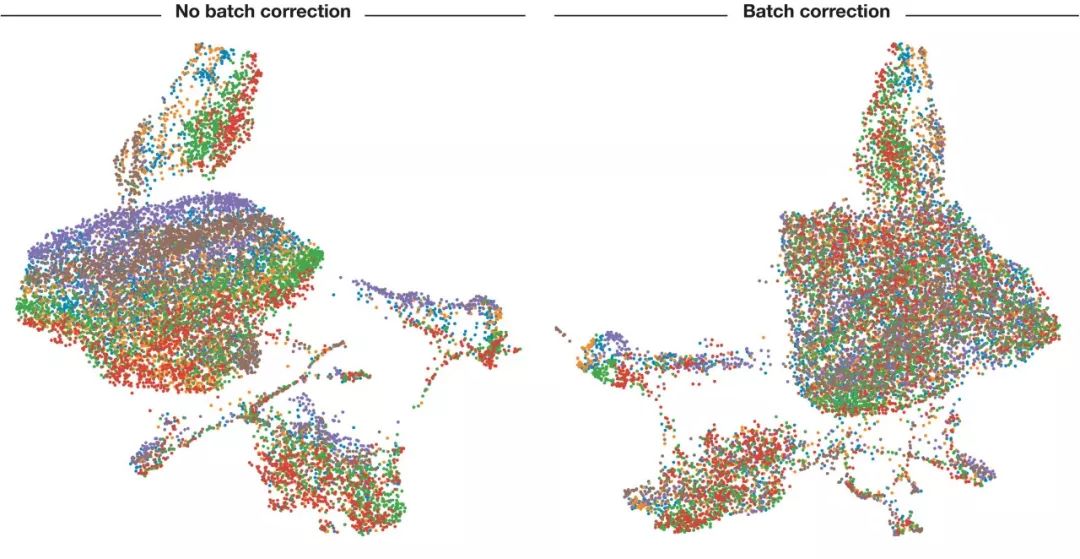
图3. 批次校正前后UMAP可视化结果展示
细胞按其来源的样本上色。在批次校正前可以看到颜色区分很明显(细胞所来源的样品对细胞分群影响大),表明批次影响比较大。使用ComBat校正批次后,同一个颜色分布比较散了,细胞所来源的样品对细胞分群影响变小了。
与批次校正相比,数据整合面临的另一个挑战是数据集之间的组成差异 (compositional differences)。估计批次效应时,ComBat使用同一批次的所有细胞来拟合批次校正参数。这种方法将批次效应与数据集之间非共有的细胞类型或状态之间的生物学差异混淆。数据整合方法,例如典型相关分析(Canonical Correlation Analysis, CCA),相互最近邻(Mutual Nearest Neighbours, MNN), Scanorama, RISC, scGen, LIGER, BBKNN和Harmony已经开发用于解决这个问题。尽管数据整合方法也可应用于简单的批次校正处理,但是鉴于非线性数据整合方法会增大自由度,使用时需要注意过度校正问题。例如,在较简单的批次校正设置中,ComBat的表现优于MNN(Buttner et al.,2019)。需要进一步进行数据整合和批次校正方法之间的比较研究,才可以评估这些方法的适用范围。
缺失值填充
技术数据校正的另一种类型是缺失值填充(也称为降噪或插补, denoising or imputation)。单细胞转录组的数据包含各种噪声。这种噪音的一个特别突出的来源是dropout。推断dropouts事件,用推断出的合适的表达值替换这些零以减少数据集中的噪声成为几种最新工具的目标 (MAGIC, DCA, scVI, SAVE, scImpute)。已证明进行缺失值填充可改善基因与基因相关性的估计。此外,这一步也可以与标准化、批次校正和其他下游分析整合,就像在scVI工具中实现的那样。尽管大多数数据校正方法都将标准化后的数据作为输入,但是某些缺失值填充方法是基于预期的负二项噪声分布,需要基于原始计数矩阵进行操作。在应用缺失值填充时,应考虑到没有一种方法是完美的。因此,任何方法都可能会对数据中的噪声进行过高校正或校正不足。确实,已有报道表明缺失值填充可能引入错误的相关信号。鉴于在实际应用中难以评估缺失值填充是否得当,用户选择是否应用这一方法也是很大的挑战。当前缺失值填充方法是否能拓展应用到大数据集还是一个问题。鉴于这些考虑,目前尚无关于应如何使用缺失值填充的共识。谨慎的方法是仅在视觉展示数据时使用缺失值填充,而非在探索性数据分析过程中基于填充的数据作出推论或假设。全面的实验验证在这里尤为重要。
陷阱和建议:
- 仅在进行轨迹推断和校正的生物学混杂因素不影响感兴趣的生物学过程时才校正这些因素的影响。
- 如果校正的话,所有因素同时校正而不是分别校正技术和非关注的生物因素变量。
- 基于板的数据集预处理时需要校正
count数的影响,建议采用非线性标准化方法或downsampling方法进行标准化。- 当批次之间的细胞类型和状态组成一致时,建议通过
ComBat执行批次校正。- 数据整合和批次校正应通过不同的方法进行。数据整合工具可能会过度校正简单的批次效应。
- 用户需要对只在缺失值填充后才能发现的信号格外注意。探索性分析时最好不进行缺失值填充操作。
特征选择、降维和可视化
人单细胞RNA-seq数据集可包含多达25,000个基因的表达值。对于一个给定的scRNA-seq数据集,其中有许多基因都不能提供有用信息,并且大多只包含零计数。即使在QC步骤中滤除了这些零计数基因后,单细胞数据集的特征空间也可能超过15,000个维度(即还会剩余15,000多基因)。为了减轻下游分析工具的计算负担、减少数据中的噪声并方便数据可视化,可以使用多种方法来对数据集进行降维。
特征选择
scRNA-seq数据集降维的第一步通常是特征选择。在此步骤中,对数据集基因进行过滤仅保留对数据的变异性具有信息贡献的基因(在数据中变异大的基因)。这些基因通常被定义为高变化基因(HVG,highly variable genes)。根据任务和数据集的复杂性,通常选择1,000到5,000个HVG用于下游分析。Klein et al.的初步结果表明,下游分析对HVG的数量不太敏感。在HVG数量从200到2,400之间选择不同的数目时,评估显示PCA结果相差不大。基于此结果,我们宁愿选择更多的HVG用于下游分析 (err on the side of:可以借鉴的英语短语)。
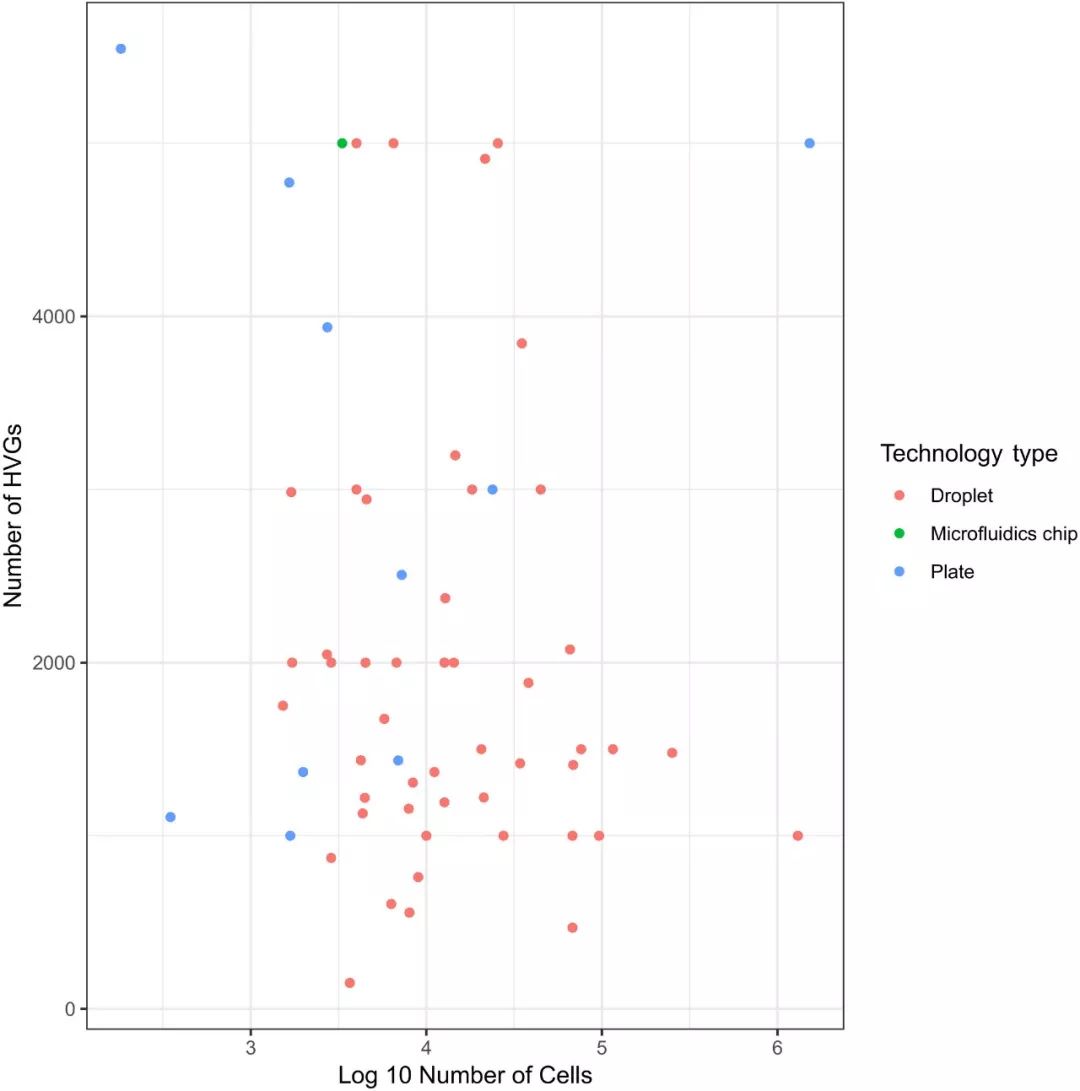
图EV1. 不同大小数据集选择HVG基因数量的分布展现。(数据来源于手动整理的最近发表的多篇scRNA-seq文章,颜色代表不同的技术方式)
在Scanpy和Seurat中都实现了一种简单而流行的选择HVG的方法。在这里,基因按其均值表达进行分组,将每个组内方差/均值比最高的基因选为每个分组的HVG。该算法在不同软件中输入不同,Seurat需要原始count data;Cell Ranger需要对数转换的数据。最佳地,应在技术等干扰因素校正之后选择HVG,以避免选择仅由于例如批次效应等引入的高可变基因。Yip et al.综述了其他HVG选择方法。
特征选择后,可以通过专用的降维算法进一步对单细胞表达矩阵进行降维。这些算法将表达式矩阵映射到低维空间中,同时以尽可能少的维数捕获数据中所有的信息。鉴于单细胞RNA测序数据固有的低维性特征,这一方法是合适的。也就是说,细胞表达图谱构成的生物形态 (biological manifold)可以使用远少于基因数目的维度信息来展示。降维旨在找到这些维度。
降维有两个主要目标:可视化和信息汇总(summarization)。可视化是尝试在二维或三维空间最优地展示数据集。降维后的维度值就是数据在新的空间进行可视化如绘制散点图时的坐标值。信息汇总没有规定输出的维数;但更高的维数对表示原有数据的差异越来越不重要(生信宝典注:可以理解为PCA中各个主成分对于原始数据差异的解释依次降低)。汇总技术可通过计算数据的固有维数来将数据降维到基本组成(主)成分,从而有助于下游分析。虽然不应使用二维可视化数据来汇总数据集,但汇总方法获得的降维数据可用来可视化数据。另外,专用的可视化技术通常会更好地展示数据的原始结构和变异性 。
降维后的维度是通过对基因表达向量进行线性或非线性组合生成的。特别是在非线性情况下,降维后的数据难以解释其生物含义。图4中显示了一些常用降维方法的示例应用。随着可供选择的方法的增加,详细讨论这些方法已超出了本教程的范围。相反,我们简要概述了可能有助于用户在常见的降维方法之间进行选择时需要考虑的因素。Moon et al. 提供了有关单细胞分析降维的更详细的综述 (Manifold learning‐based methods for analyzing single‐cell RNA‐sequencing data. Curr Opin Syst Biol 7: 36–46)。
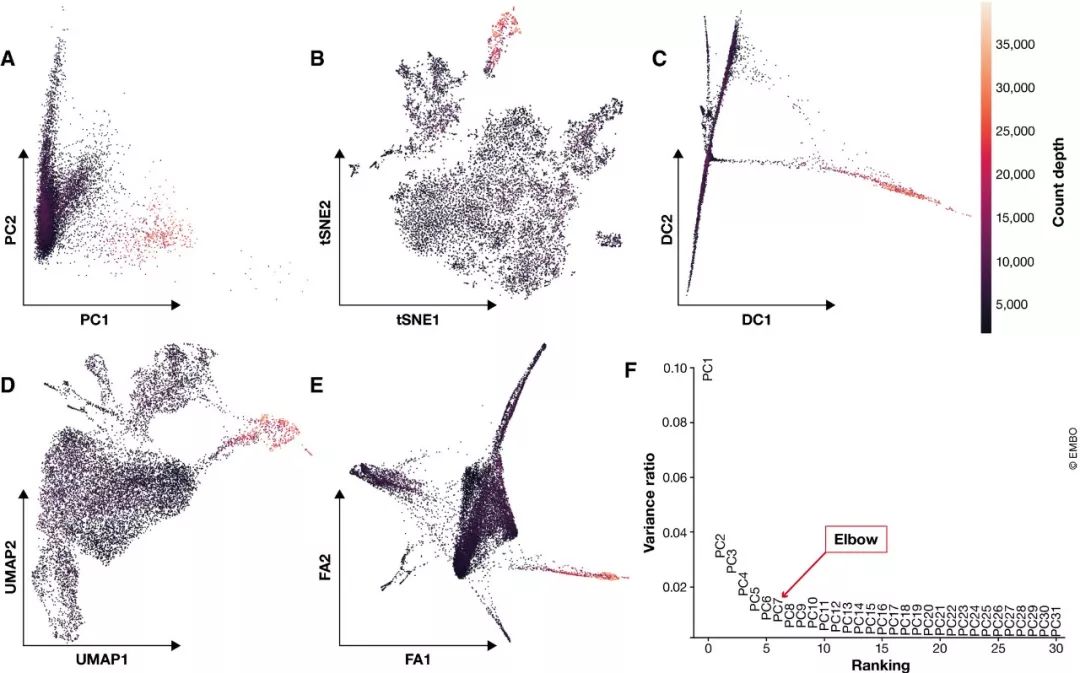
图4. scRNA‐seq数据常用的可视化方法。
(A) PCA, (B) t‐SNE, (C) diffusion maps, (D) UMAP and (E) A force‐directed graph layout via ForceAtlas2. 细胞根据测序深度上色 (F) 前31个主成分解释的原始数据的差异。图中的拐点( “elbow”)用于选择下游分析相关的主成分,位于PCs 5 和PCs 7之间。
主成分分析(PCA)和diffusion maps是两种常用的降维方法,在单细胞分析中也很流行。主成分分析是一种线性方法,通过最大化每个可能维度中捕获的残差 (residual variance)来进行降维。尽管PCA不能像非线性方法那样在更少的维度捕获原始数据更多的信息,但是它是许多当前可用的聚类或轨迹推断工具的基础。实际上,PCA通常用作非线性降维方法的预处理步骤。通常,PCA通过其前N个主成分来代表原始数据集,其中N可以通过elbow算法(参见图4F)或基于置换检验的jackstraw方法确定。PCA简单线性化的优势是:降维空间中的距离在该空间的所有区域具有一致的解释。因此,我们可以将感兴趣的统计量与主成分进行关联分析,以评估其重要性。例如,可以将主成分投影到技术干扰协变量上,以研究QC、数据校正和标准化过程的效果(Buttner et al.,2019),或显示基因在数据集中的重要性 。diffusion map是一种非线性数据降维技术。由于diffusion component强调数据的转换,因此主要用于诸如细胞分化之类的连续过程。通常,每个diffusion component(即diffusion map 维度)突出显示不同细胞群体的异质性。
可视化
可视化时一般使用非线性降维方法(图4)。scRNA-seq数据可视化的最常见的降维方法是t‐SNE ( t‐distributed stochastic neighbour embedding) 。t‐SNE的维度着重于以牺牲全局结构为代价来保留局部相似性 (生信宝典注:PCA则是尽可能多的保留全局差异)。因此,这些可视化可能会夸大细胞群体之间的差异,而忽略群体之间的潜在联系 (t‐SNE dimensions focus on capturing local similarity at the expense of global structure. Thus, these visualizations may exaggerate differences between cell populations and overlook potential connections between these populations)。另一个困难是对参数perplexity parameter的选择,因为t-SNE图会因为这个参数值不同而显示出明显不同的分簇数。t‐SNE的常见替代方法是UMAP (Uniform Approximation and Projection method)或基于图的工具,例如SPRING。UMAP和SPRING的力导向布局算法ForceAtlas2(force‐directed layout algorithm)可以说是数据潜在拓扑结构展示最好的方法(Wolf et al,2019)。在此比较中,使UMAP与众不同的是它的速度快和能应用于更大规模数据的能力(Becht et al,2018)。因此,在没有特定生物学问题限制的情况下,我们将UMAP视为探索性数据可视化分析的最佳实践。此外,UMAP还可以把数据降维到二维以上的新数据。尽管我们不知道UMAP`在数据汇总中的任何应用,但它可能是PCA的一个合适的替代方案。
在细胞水平上经典可视化方法的替代方法是PAGA (partition‐based graph abstraction)。事实证明,此工具可以充分展示数据的拓扑结构,同时使用聚簇生成粗粒度的可视化图像。结合以上任何一种可视化方法,PAGA可以生成粗粒度的可视化图像 (coarse‐grained visualizations),从而可以简化单细胞数据的解释,尤其是在测序细胞量大或整合了大量细胞的情况下。
陷阱和建议:
- 我们建议根据数据集的复杂程度选择
1,000至5,000个高可变基因 (HVG)。- 当将基因表达值归一化为
均值为0和单位方差时,或者将模型拟合的残差用作标准化表达值时,不能使用基于基因表达均-方差的特征选择方法。因此,在选择HVG之前,必须考虑要执行哪些预处理。- 信息汇总 (
summarization,类比于挑选重要的主成分)和可视化应使用不同的降维方法。- 我们建议使用UMAP进行探索性分析可视化;使用PCA做为通用数据降维方法;
diffusion maps可以在轨迹推断时替代PCA。- UMAP+PAGA是可视化特别复杂的数据集的合适替代方法。
数据预处理步骤
虽然我们在上面按顺序概述了scRNA‐seq中常见的预处理步骤,但下游分析通常需要不同级别的预处理数据,建议根据下游应用调整预处理的各个步骤。为了向新用户说明这种情况,我们将预处理分为五个数据处理阶段:(i)原始数据,(ii)标准化后的数据,(iii)校正后的数据,(iv)特征选择后的数据,以及(v)降维后的数据。数据处理的这些阶段可以归类为三个预处理层:测量的数据,校正的数据和降维的数据。细胞和基因的质量控制筛选是必须执行的步骤,因此在此处略过。尽管层的顺序代表了scRNA-seq分析的典型工作流程,但也可以跳过某一步或在处理阶段的顺序中稍作更改。例如,单批次数据集可能不需要批次校正。在表1中,我们总结了预处理数据的每一层所适用的下游处理程序。
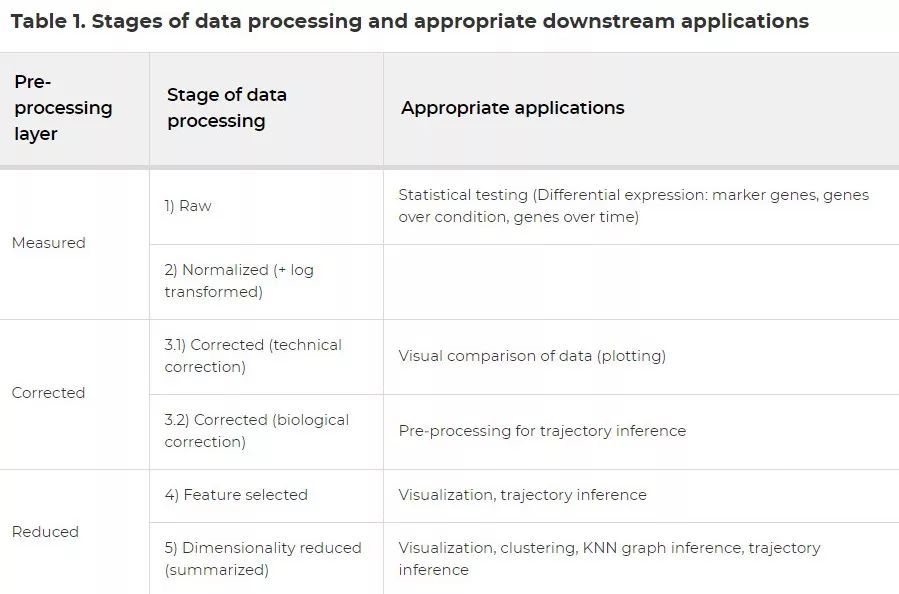
表1
表1中的预处理阶段分为三组:测量的数据,校正后的数据和降维后的数据。我们将测量的数据定义为原始数据和处理后但保留了表达值为零的基因的数据。应用细胞特异的size factor的全局标准化方法即使在log(x+1)转换后仍保留零表达值。相比之下,校正后的数据会去除不需要的变化信息进而替换零表达值。校正后的数据层表示数据的“最干净”版本,它是数据代表的生物信号的最近似值。我们称最终预处理层为降维后的数据(reduced data)。此数据层强调数据的主要差异,可以使用降维后的特征集来描述。
前述特性决定了预处理数据对特定下游应用的适用性。作为最后的预处理阶段,降维后的数据是广泛应用的数据层。但是,差异表达分析只能在基因空间内进行生物学解释,而无法(或无法完全)体现在降维后的数据中。降维后数据的优势在于生物数据信息的汇总(summarization)和影响生物数据的噪声的降低。因此,降维后的数据应用于后续的探索性方法(如可视化、邻域图推断、聚类)以及计算复杂的下游分析工具(如轨迹推断)。实际上,许多轨迹推断方法在工具本身中都包含了降维功能。
单个基因的表达谱只能在基因空间中进行比较,这一信息存在于测量的数据和校正后的数据中。表达谱可以进行视觉和统计学比较。我们认为视觉比较和统计比较应该在不同的数据层上进行。对于目测基因表达,校正的数据是最合适的,有助于结果解释。如果对原始数据进行视觉比较,则要求用户牢记数据中的偏差以准确解释结果。但是,此处应分别考虑技术和生物协变量的校正数据。虽然对生物协变量的校正可能会增加特定生物信号的强度,但其对潜在的生物学意义表示可能不甚准确,并且会掩盖可能相关的其他生物信号。因此,经过生物学不相关因素校正的数据主要适用于专注于特定生物学过程(例如轨迹推断方法)的分析工具。
基因表达的统计比较需要基于测量数据层。去噪、校正批次或其他噪声源的方法总不是太完美。因此,数据校正方法不可避免地会对数据进行过度或不足校正,因此会以意想不到的方式改变至少某些基因表达谱的方差。基因表达的统计检验依赖于将背景方差评估为数据噪声的空模型。随着数据校正趋于减少背景变化(图EV2),背景变化被数据校正方法过度校正的基因将更有可能被评估为显著差异表达基因。此外,某些数据校正方法(例如ComBat)将不吻合于实验设计的表达信号定义为噪声,随后将其从数据中删除。这一基于实验设计的优化方法除了可能造成数据中的噪声被低估还会导致对效应大小 (effect size)的高估。鉴于这些考虑,与校正后的数据相比,基于测量数据进行差异检验是一种更为保守的方法。使用测量的数据时,可以并且应该在差异统计分析模型中考虑技术协变量 。
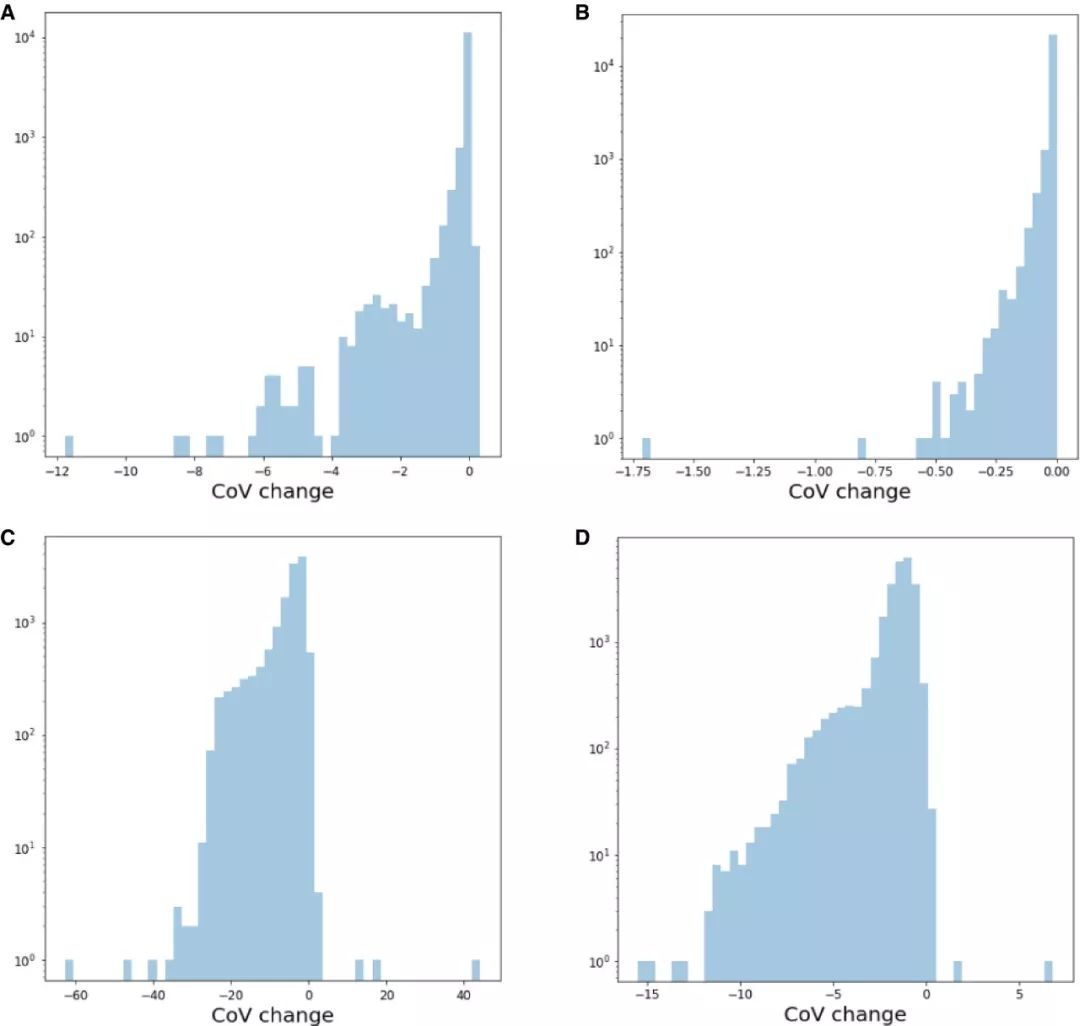
图EV2. 批次校正和降噪后变异系数的变化
负值代表数据校正后变异系数降低。第一行展示的是ComBat校正(A) mouse intestinal epithelium (mIE) 和 (B) mouse embryonic stem cell (mESC) 数据变异系数的变化。第二行展示的是DCA降噪后(C) mIE 和 (D) mESC 数据变异系数的变化。
最近一篇单细胞差异表达分析方法的文章也支持以上观点, 它只使用原始或标准化后的矩阵作为输入 (Soneson & Robinson, 2018)。这篇研究中标准化后的数据仅限于全局标准化方法。然而当前可用的非线性标准化方法模糊了标准化和数据校正的界限(具体见标准化部分)。这样标准化之后的数据已不适合用于差异基因分析。
陷阱和建议:
- 原始测量的数据用于差异基因统计检验分析;
- 校正后的数据用于数据的可视化比较;
- 降维后的数据用于其他基于数据形态(
biological data manifold)的下游分析。
下游分析
经过预处理后,我们称之为下游分析的方法指应用于生物学发现并描述潜在的生物学系统的方法。通过将可以解释的模型拟合到数据中获得相应的结论,比如有相似基因表达谱的细胞群代表一个细胞簇、相似细胞之间基因表达的微小变化指示连续(分化)轨迹;或具有相关表达趋势的基因指示共调控等。
下游分析可分为细胞水平和基因水平的方法,如图5所示。细胞水平分析通常着重于两种结构的描述:簇和轨迹。这些结构又可以在细胞和基因水平上进行分析,即簇分析和轨迹分析方法。广义地讲,簇分析方法试图将细胞分类为离散的多个组来解释数据的异质性。相反,在轨迹分析中,数据被视为细胞连续变化动态过程的一个个快照,轨迹分析方法研究了这一连续变化过程。
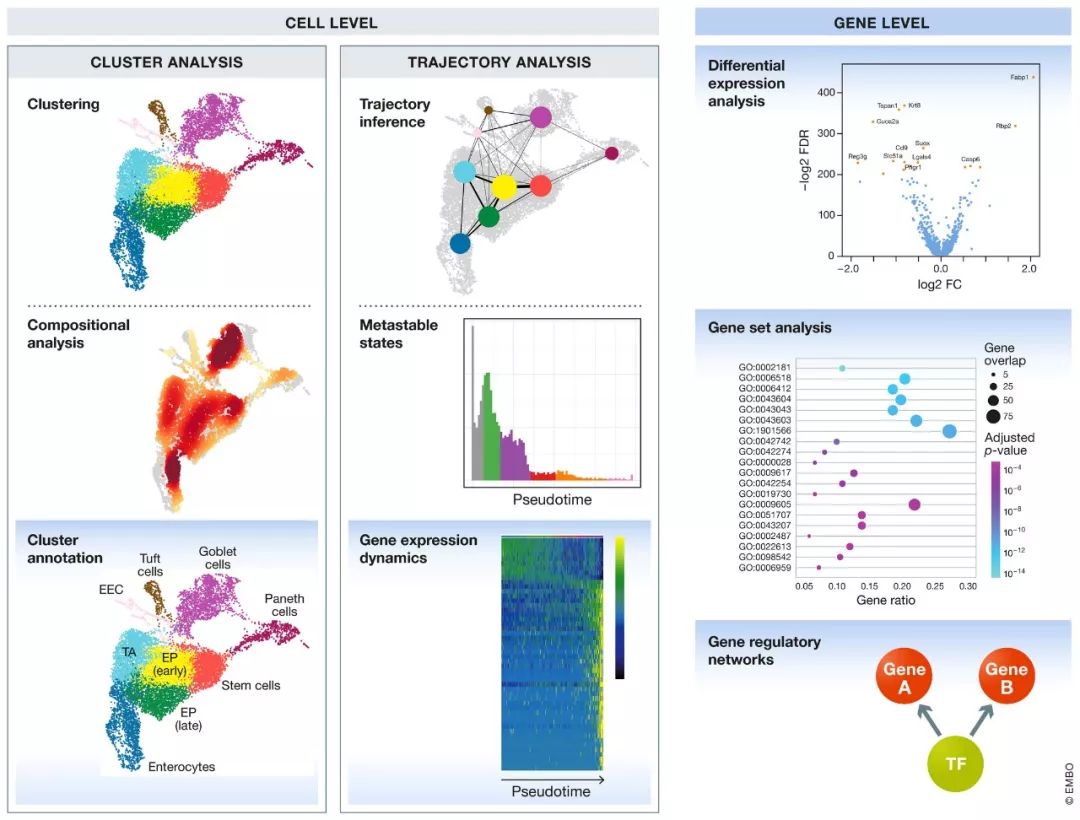
图5. 下游分析方法总览(蓝色背景的方法是基因水平的分析方法)。
在此,我们在详细描述独立于这些细胞结构进行的基因水平分析之前,先描述细胞水平和基因水平的聚类和轨迹分析工具。
聚类分析
聚类。将细胞聚类成簇通常是任何单细胞分析的第一个中间结果。聚类成簇使我们可以推断成员细胞的身份。簇是通过基于细胞基因表达谱的相似性将细胞分组得到的。表达谱相似性是通过对将降维的数据进行距离度量确定的。相似性评分的一个常见示例是在主成分降维后的表达空间上计算的欧氏距离。存在两种根据这些相似性分数生成细胞簇的方法:聚类算法和基于图的社群检测方法 (community detection methods)。
聚类是直接基于距离矩阵的经典无监督机器学习问题。通过最小化簇内距离或在降维后的表达空间中鉴定密集区域,将细胞分配给簇。流行的k均值聚类算法通过确定聚类质心并将细胞分配给最近的聚类质心,将细胞分为k个类。质心位置经过迭代优化。这种方法需要输入预期的簇数,但这通常是未知的,必须进行启发式校准。k均值在单细胞数据中的应用因所使用的距离度量而异。标准距离度量是使用欧氏距离,替代距离包括余弦相似度、基于相关的距离度量或SIMLR方法,该方法使用Gaussian kernels为每个数据集计算距离度量。最近的比较表明,与k均值一起使用或作为Gaussian kernels分析的基础时,基于相关的距离可能会胜过其他距离度量方法。
社群检测方法是一种图分类算法,依赖于单细胞数据的网络图表示。图是使用K最近邻方法(KNN图)获得的。细胞在图中表示为节点。每个细胞与其K个最相似的细胞相连,通常对主成分降维后的数据计算欧氏距离作为相似性度量。根据数据集的大小,K通常设置为5到100个最近的邻居。获得的KNN结果图捕获了表达图谱的基础拓扑结构(Wolf et al., 2019)。表达谱相似的细胞集合对应于图的紧密连接区域,然后使用社群检测方法检测图中这些紧密连接区域。社群检测方法通常比聚类方法要快,因为只有相邻的细胞对会被视为属于同一群集。因此,这种方法大大减少了可能的细胞簇的搜索空间。
在开创性的PhenoGraph方法(Levine et al.,2015)出现之后,聚类单细胞数据集的标准方法已演变成多分辨率模块优化算法 (multi‐resolution modularity optimization),即在单细胞KNN图中应用Louvain算法。此方法是在Scanpy和Seurat单细胞分析平台中应用的默认聚类方法。评估发现,这一方法应用于单细胞RNA测序数据以及流式细胞和质谱数据分析时优于其他聚类方法。从概念上讲,Louvin算法将组内细胞连接数高于预期的一组细胞视作一个簇。(生信宝典注:Louvain algorithm算法采用迭代方式,先计算每个点加入到其邻居节点所在社区带来的模块性评估收益,然后将其加入收益最大的邻居节点所在社区,对所有未归类点重复进行这一过程,直至结果稳定。然后再把每个社区作为一个节点,重复上一步。) 优化后的模块鉴定函数包括一个分辨率参数,该参数允许用户确定簇的近似大小。通过对KNN图取子集,还可以只聚类一部分特殊的细胞簇。这种子集聚簇模式可以允许用户识别某个细胞类型簇内的细胞状态,但也可能会发现仅由数据中的噪声带来的聚类模式。
陷阱和建议:
- 我们建议在单细胞KNN图上执行Louvain社群检测算法获得聚类簇。
- 细胞聚簇不一定使用单一分辨率进行。对特定细胞簇进行细化聚类是鉴定数据集中亚结构的有效方法,可以发现细胞亚群。
细胞簇注释
在基因水平上,对每个簇的分析是鉴定其标记基因 (Marker genes)。这些所谓的标记基因代表了细胞簇的特征,用于给细胞簇一个有生物学意义的标签。该标签表示集群中细胞的身份。由于任何聚类算法都会聚类出细胞簇,因此聚类获得的生物簇的准确性只能通过其生物学注释进行衡量 (生信宝典注:这也是前面和易生信课程中反复强调的,细胞过滤时标准尽量松一些,根据聚类结果回看之前的参数设置是否合理。)。
尽管把单细胞数据中鉴定到的簇归类为一个有生物意义的细胞类型是一个诱人的结果,但是细胞身份的确定不是那么简单(Wagner et al., 2016;Clevers et al., 2017)。首先,并不总是清楚细胞类型定义的精确程度。例如,“T细胞”可能是某些人满意的细胞类型标记,但其他人可能会在数据集中寻找T细胞亚型并区分CD4+T细胞和CD8+T细胞 。另外,同一细胞类型的不同状态可能会被分到不同的细胞簇。基于上述原因,最好使用术语cell identities而不是cell types。在对细胞进行聚类和注释之前,用户必须确定要注释到的细胞信息精确水平,从而确定合适的聚簇分辨率。
识别和注释细胞簇依赖于外部信息,这些信息描述了每个细胞类群预期的表达谱。随着小鼠脑图集(Zeisel et al., 2018)、人类细胞图集(Regev et al., 2017)和其他工作的继续,可用的参考数据库越来越多。这些数据库极大地方便了细胞簇注释。在没有相关参考数据库的情况下,可以通过将细胞聚类簇的Marker基因与文献中的Marker基因进行比较来注释细胞身份(请参阅Github上的案例研究)或通过直接对文献报道的标记基因在自己研究的数据中进行表达值可视化来确定细胞身份(图6B)。应当注意的是,后一种方法将用户限制于普通转录组数据鉴定出的经典细胞类型,而不是对细胞身份 (cell identities)的理解。而且,研究表明常用的细胞表面标志基因在定义细胞身份上是效果有限的(Tabula Muris Consortium et al., 2018)。
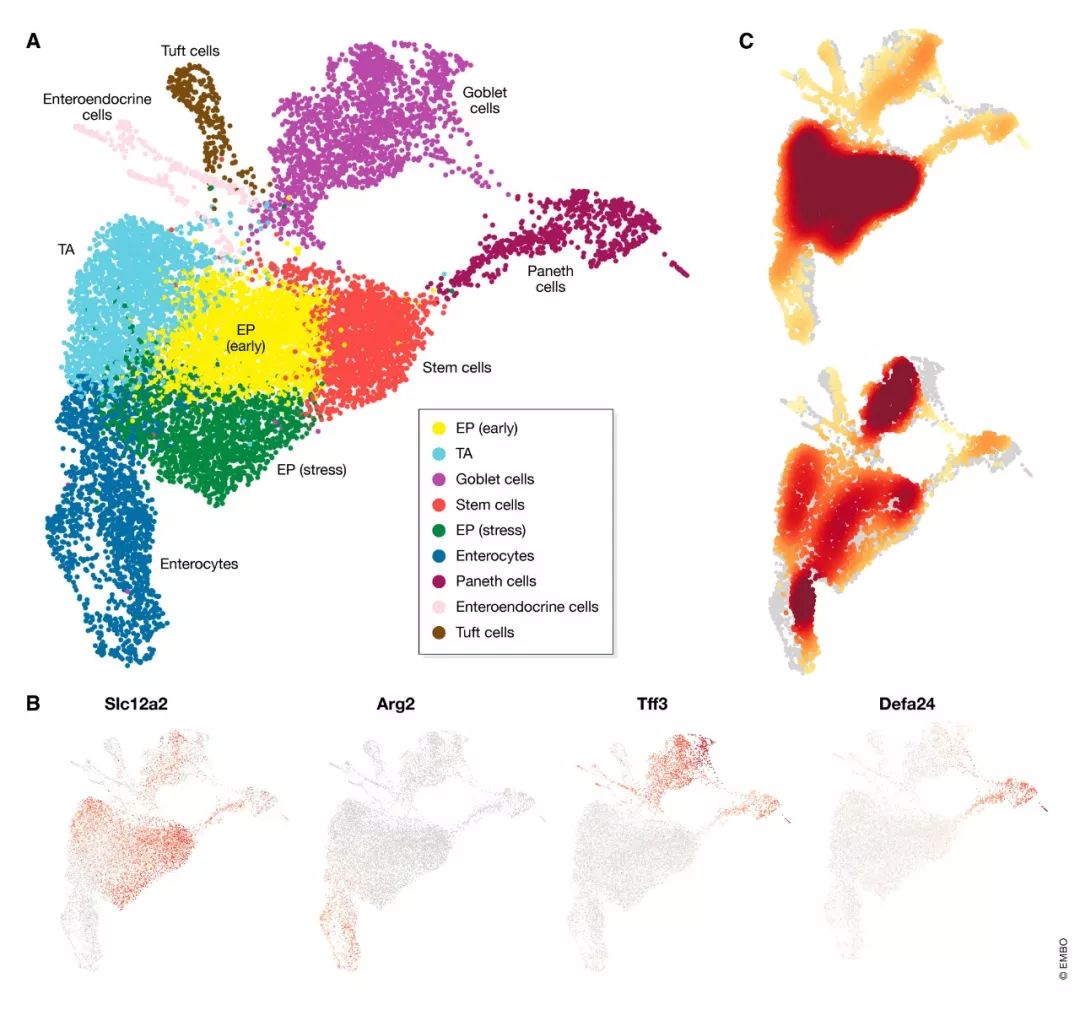
图6. 聚类分析结果
(A) UMAP可视化Louvain算法鉴定的细胞簇,并标记注释的细胞类型名称。(B) 通过细胞类型Marker基因的表达标记细胞簇所代表的细胞类型,如干细胞 (Slc12a2), 肠上皮细胞 (Arg2), 杯状细胞 (Tff3) 和潘氏细胞 (Defa24)。灰色代表低表达,红色代表高表达。Marker基因也可能在其它细胞簇有表达,比如Tff3在杯状细胞和潘氏细胞都有表达,只是在杯状细胞更高。(C) 肠上皮细胞区域细胞类型组成热图(上面是近端区域,下面是远端区域)。相对细胞密度高的区域用深红色表示。
有两种方法可以使用参考数据库信息注释细胞簇:使用计算出的Marker基因或使用完整的基因表达谱。可以通过对目标簇的细胞和数据集中的所有其他细胞进行差异表达(DE)分析来鉴定标记基因集。通常,我们关注在目标簇中上调的基因。由于预期标记基因表达变化幅度较大,因此通常使用简单的统计检验(例如Wilcoxon秩和检验或t检验)就可以对两组细胞的基因进行差异检验分析。根据统计检验结果,选择排名最高的基因视为标记基因。可以通过比较自己的数据中鉴定出的标记基因和来自参考数据集的标记基因对细胞簇进行注释。一些在线工具如www.mousebrain.org (Zeisel et al, 2018) 或http://dropviz.org/ (Saunders et al, 2018)允许用户在参考数据集中可视化用户鉴定出的Marker基因从而方便细胞簇的注释。
鉴定标记基因时应注意两个方面。首先,用于鉴定标记基因的P值基于以下假设:即鉴定出的细胞簇是有生物学意义的。如果细胞簇鉴定过程中存在不确定性,则必须在统计检验中考虑簇分配与标记基因鉴定之间的关系。这个关系起因于鉴定细胞簇和标记基因都是基于同一套基因表达数据。差异基因检测的零假设(null hypothesis)是两组细胞整体基因的表达值具有相同的分布。然而,由于这两个聚类组是基于基因表达变化的聚类结果得到的,其基因表达谱从本质上肯定存在差异。因此即使在应用splatter随机生成的数据的聚类结果中也可以鉴定出显著的Marker基因 (Zappia et al, 2017)。为了在聚类数据中获得合理的显著性度量,可以使用置换检验减少聚类步骤带来的影响。补充介绍S3中对这一检验有详细描述。最近的一个差异表达工具也专门解决了这个问题(Preprint:Zhang et al,2018)。
在当前的研究中,P值通常会被夸大,这可能导致对标记基因数量的高估。但是,基于P值对基因进行排序则不受影响。假设聚类结果是有生物学意义的,那么排名最高的标记基因仍将是最佳候选标记基因。首先,我们可以通过可视化展示粗略地查验获得的标记基因。我们强调,特别是通过无监督聚类方法定义细胞聚类簇时,会导致夸大的P值。当改为通过单个基因的表达确定细胞簇的身份时,P值可以解释为相对于其他基因的期望值。尽管很常见,但是这种单变量的聚类簇注释方法除了在特定情况下不建议使用(例如,β细胞中的胰岛素或红细胞中的血红蛋白)。其次,标记基因在数据集中将一个细胞簇与其他细胞簇区分开,不仅依赖于细胞簇鉴定结果,还依赖于数据集的组成。如果数据集组成不能准确表示背景基因表达,则检测到的标记基因将偏向在其它细胞中未检测到的基因。特别是在细胞多样性低的数据集中计算标记基因时,必须特别考虑这一方面。
最近,自动细胞簇注释已可用。通过直接将注释好的参考簇的基因表达谱与单个细胞进行比较,使用诸如scmap(Kiselev et al., 2018b)或Garnett(Preprint:Pliner et al., 2019)之类的工具可以在参考集和自己的数据集之间比较注释。因此,这些方法可以同时执行细胞注释和细胞簇鉴定,而无需数据驱动的细胞聚类步骤。但由于不同实验条件下细胞类型和状态组成不同(Segerstolpe et al.,2016;Tanay&Regev,2017),基于参考数据的聚类不应取代数据驱动的聚类过程。
聚类、聚类注释、重新聚类或子聚类以及重新注释的迭代是很耗时的过程。自动化的细胞簇注释方法大大加快了此过程。但是,自动和手动方法各自存在优点和局限性,速度的提高与灵活性的降低并存,因此很难有一种全局适应的方法。如前所述,参考数据集很难与正在研究的数据集包含完全一样的细胞类型。因此,不应放弃用于手动注释标记基因的计算。特别是对于包含许多细胞簇的大型数据集,当前的最佳实践是两种方法(自动+手动)的组合。为了提高速度,可以使用自动化细胞类型注释工具粗略地标记细胞并确定可能需要鉴定细胞子群的簇。随后,应针对细胞簇计算标记基因,并将其与参考数据集或文献中的已知标记基因集进行比较。对于较小的数据集和缺少参考数据库的数据集,手动注释就足够了。
陷阱和建议:
- 不要使用标记基因的P-value值来验证细胞簇身份,尤其是当检测到的标记基因无益于注释细胞簇时。P值可能会被夸大。
- 请注意,由于数据集的细胞类型和状态组成,同一细胞簇的标记基因可能在数据集之间有所不同。
- 如果存在相关的参考数据集,我们建议综合使用自动聚类注释和基于数据的标记基因的手动注释来注释细胞类型。
细胞组成分析 (Compositional analysis)。在细胞层面,我们可以根据其组成结构分析聚类数据。细胞组成数据分析围绕不同样品落入每个细胞簇的细胞比例进行分析。这一比例可能在疾病状态下发生变化。例如,研究表明沙门氏菌感染会增加小鼠肠上皮区域肠上皮细胞(enterocytes)的比例(Haber et al.,2017)。
要研究单细胞数据的细胞组成变化,需要足够的细胞数量来稳健地评估细胞簇的比例,并需要足够的样本数量来评估细胞簇组成中的背景变化。由于合适的数据集直到最近才可用,因此专用工具尚待开发。在上述小鼠研究中,细胞身份计数使用泊松分布建模,使用实验条件作为协变量和检测到的细胞总数作为偏移量。在此,可以对回归系数进行统计检验,以评估特定细胞群体的出现频率是否发生了显著变化。但是,对同一数据集中其他细胞簇的统计检验分析并非彼此独立。如果一个细胞聚类簇的比例发生变化,那么所有其他细胞聚类簇的比例也会发生变化。因此,使用该模型无法评估整体构成是否发生了显着变化。在没有专用工具的情况下,细胞构成数据的可视化展示会给不同样品之间细胞组成变化提供有用的信息(图6C)。该领域的未来发展可能会借鉴质谱流式细胞技术(Mass Cytometry)(例如Tibshirani et al., 2002;Arvaniti&Claassen., 2017;Lun et al., 2017;Weber et al., 2018)或微生物组的方法(Gloor et al., 2017),组成数据分析在这些领域已经受到了更多关注。
陷阱和建议:
- 统计检验分析时需要考虑到样本之间的细胞簇比例的变化是互相依赖的(非独立的)
Trajectory analysis
轨迹推断。细胞多样性不能通过离散的分类系统(例如细胞聚类)充分描述。驱动观察到的细胞异质性发展的生物进程是一个连续过程(Tanay&Regev,2017)。因此,为了捕获细胞身份之间的过渡状态、不同的分化分支或生物学功能的渐进式非同步变化,我们需要动态的基因表达模型。这类方法称为轨迹推断(trajectory inference,TI)。
轨迹推断方法将单细胞数据视为连续过程的一个个快照。这一过程通过最小化相邻细胞之间的转录改变构建细胞空间的转换路径(图7A和B)。这些路径上的细胞排序由伪时间变量 (pseudotime variable)描述。虽然此变量是基于距离根细胞的转录距离计算的,但通常被解释为发育时间的代名词(Moignard et al.,2015; Haghverdi et al.,2016; Fischer et al.,2018; Griffiths et al.,2018)。
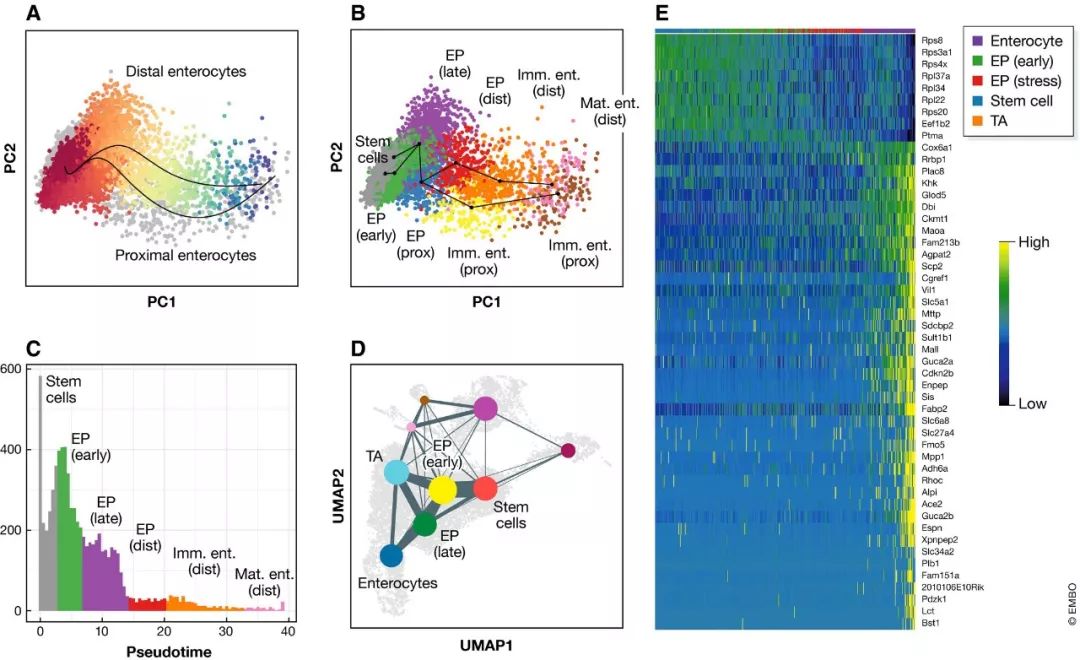
图7. 小鼠肠上皮细胞轨迹分析结果展示
(A) Slingshot鉴定出的远端和近端肠上皮细胞分化轨迹。远端谱系细胞按pseudotime从红色到蓝色上色。数据集中其它细胞用灰色点展示。(B) PCA空间中Slingshot 轨迹和细胞簇比较展示。簇名字简写如下:cuEP—enterocyte progenitors; Imm. Ent.—immature enterocytes; Mat. Ent.—mature enterocytes; Prox.—proximal; Dist.—distal. (C) 远端肠上皮细胞轨迹中每个pseudotime对应的细胞密度分布。柱子按每个pseudotime 区间主要的细胞簇上色。(D) 数据集投射到UMAP空间的抽象图展示。每个簇用不同颜色点展示。出现在其它轨迹中的簇特别标记处理用于比较分析。TA代表短暂增殖下拨 (transit amplifying cells)。(E)R包GAM展示肠上皮细胞中基因表达随pseudotime的动态变化。
自Monocle(Trapnell et al. 2014)和Wanderlust(Bendall et al. 2014)建立了TI (trajectory inference)领域以来,可用的TI方法数量激增。当前可用的TI方法的差别在于构建的发育轨迹模型拓扑结构复杂性不同,从简单的线性轨迹或二分支轨迹到复杂树形轨迹、多分支轨迹或组合多种拓扑结构轨迹。在最近对TI方法进行的全面比较中(Saelens et al.,2018)发现没有一种单独的方法可以在所有类型的轨迹分析中都表现最优 。相反,应根据预期轨迹的复杂性选择TI(轨迹分析)方法,研究比较表明Slingshot(Street et al.,2018)在简单轨迹分析如从线性轨迹到二分支和多分轨迹表现最佳。如果预期数据对应更复杂的轨迹,作者建议使用PAGA(Wolf et al.,2019)。如果知道精确的轨迹模型,则可以选择使用更特定的方法来提高性能(Saelenset al.,2018)。通常,应使用多种方法来确定评估推断出的轨迹,以避免方法偏差。
在典型的分析流程中,轨迹推断(TI)方法应用于降维后的数据。如果使用的TI工具自带了降维功能,则基于校正后的数据进行分析。由于通常在细胞内同时发生多种生物学过程,因此消除其他生物过程的影响对鉴定预期轨迹可能很有用。例如,T细胞在成熟过程中可能会经历细胞周期转换(Buettner et al.,2015)。此外,由于几种性能最好的TI方法依赖于聚类后的数据,因此TI通常在聚类之后执行。轨迹中的细胞簇可能表示稳态或亚稳态细胞(请参见“亚稳态”;图7B和C)。随后,可以将RNA velocities (RNA速度,或RNA表达动力学)叠加到轨迹上确认发育方向(La Manno et al.,2018)。(生信宝典注:新生转录本成熟过程中需要进行剪接操作。对于一个稳定表达的基因,总会在细胞中找到存在一定比例的未剪接的非成熟RNA形式,用于补充老的转录本的降解。如果一个基因刚被激活,短时间内将会有高比例的未成熟转录本。相反,当一个基因被抑制时,转录过程会早于转录本降解过程而被抑制,未成熟转录本的比例会降低。因此对于细胞中每个基因,未剪接的mRNA相对于剪接的mRNA的比例(RNA velocity)可以推断瞬时表达动力学,进一步推演组织内发生的细胞转变。https://www.nature.com/articles/d41586-018-05882-8)
推断的轨迹不一定要完全对应生物发育过程。首先,推断的轨迹仅表示转录相似性。很少有TI方法在其模型中包括不确定性评估(Griffiths et al., 2018)。因此,需要更多的信息来验证是否确实捕获了生物过程。这些信息可以来源于干扰实验、推断的调控基因动力学以及RNA velocity数据的支持等。
陷阱和建议:
- 我们建议使用
Saelens et al.(2018)的综述作为指南 。- 推断的轨迹不需要完全对应生物过程, 应该收集更多的证据来解释轨迹。
基因表达动力学。推断的轨迹也可能是转录噪声拟合的结果,一个排除方式是在基因水平上对鉴定出的轨迹进行进一步分析。在整个伪时间范围内连续平稳变化的基因是决定轨迹的关键基因,可用于识别潜在的生物学过程。此外,这一组与轨迹相关的基因应该包含调控相应生物进程的基因。调节基因可以帮助我们理解该生物过程是如何和为什么被触发的,并且可能是潜在的药物靶标(Gashaw et al., 2012)。
早期寻找轨迹相关基因的方法是沿轨迹在细胞簇之间进行差异基因分析(DE)。现在通过将基因表达与伪时间 (pusedotime)信息进行回归来鉴定随轨迹显著变化的基因。为了保证表达值随伪时间变量平滑变化,首先通过拟合样条曲线 (fit a spline)或其他局部回归方法(例如loess)对伪时间进行平滑处理。回归框架的区别有两点:一是其假设的噪声模型不同,二是构建的表达-伪时间函数的类别不同。通过基因对伪时间的依赖性进行模型筛选获得潜在的调控基因。这种基于伪时间回归进行的DE基因测试也会受到轨迹推断方法的影响,就像不同细胞簇之间的差异基因分析会受到聚类方法的影响一样(参见“细胞簇注释”部分)。因此,在此步骤中获得的P值不应视为显著性的评估。
当前几乎没有专用于分析基因表达时间动力学的分析工具。BEAM是整合于Monocle 轨迹推断方法中的一个工具,可以分析分支特异的基因表达动力学。除了这个,用户可以选择使用LineagePulse (https://github.com/YosefLab/LineagePulse),这个工具考虑了dropout噪音`,但仍然在开发中。或者基于标准R包或Limma包写作自己的分析测试工具。在Slingshot的使用帮助中可以找到一个这样应用的例子供参考。
由于可用的工具很少,因此尚不能确定研究基因时间表达动力学的最佳方法。使用上述所有方法肯定可以对基因表达动力学进行探索性研究。将来,高斯过程 (Gaussian processes)可能会提供一个natural model来研究基因的时间动力学。此外,对调节模块整体而不是单个基因进行统计检验分析可能会提高信噪比并更方便生物学解释。
亚稳态。轨迹的细胞水平分析是指研究伪时间轴上的细胞密度。假设以无偏方式对细胞进行随机采样,轨迹中细胞密集的区域表示优选的转录状态。当将轨迹解释为时间过程时,这些密集区域可能代表例如发育中的亚稳态(Haghverdi et al., 2016)。我们可以通过绘制伪时间坐标的直方图来找到这些亚稳态(图7C)。
细胞水平联合分析。聚类和轨迹推断代表单细胞数据的两个不同视角,并可以在粗粒度(coarse‐grained)的图形展示中进行统一。用点代表单细胞簇,轨迹作为簇之间相连的边,可以同时展示数据的静态和动态属性。这一联合分析在PAGA工具中首先提出。PAGA提出一个统计模型进行细胞簇互作分析,将细胞相似度高于期望值的两个簇之间用线连接。在最近的综述中 (Saelens et al, 2018),PAGA相比于其它轨迹分析方法整体表现更优。PAGA是唯一一个讨论到的可以处理不相连的拓扑结构和包含环形结构的复杂轨迹图的一个工具。这一特征使得PAGA在可视化整个数据集并进行探索分析时很有用。
基因水平分析。到目前为止,虽然我们专注于表征细胞结构的基因水平分析方法,但是单细胞数据的基因水平分析有更广泛的内容。差异表达分析、基因集分析和基因调控网络分析都可以直接研究数据中的分子信号。这些方法不是描述细胞异质性,而是把这种异质性作为理解基因表达差异的背景。
差异表达分析。表达数据分析的一个常见问题是两个实验条件之间是否有基因发生了差异表达。差异基因分析源于大量细胞基因表达分析(Scholtens&von Heydebreck,2005),已经有过很好的描述。相对于bulk差异分析,单细胞的优势是可以基于细胞簇进行细胞异质性评估。分析某个类型细胞簇在不同实验条件下的转录响应(Kang et al.,2018)。虽然用于解决同样的问题,普通和单细胞差异基因分析工具在方法上差别很大。普通转录组的差异基因分析方法解决的是少量样品中精确评估基因表达变化的问题,而单细胞数据中不存在这一问题。另一方面,单细胞数据包含特有的技术噪音如dropout和高细胞变异性 (Hicks et al, 2017; Vallejos et al, 2017)。这些影响因素在设计单细胞专用的差异分析方法时都要有考虑。然而,在最近一个大规模的差异基因分析方法评估中发现普通转录组差异基因检测工具与表现最好的单细胞差异分析工具性能相当 (Soneson & Robinson, 2018)。进一步地,当在普通转录组的差异分析工具中引入基因权重对单细胞数据建模时,这些工具的表现优于同类的单细胞差异分析工具 (Van den Berge et al, 2018)。基于这个比较分析,DESeq2 (Love et al, 2014) 和EdgeR (Robinson et al, 2010) 加ZINB‐wave (Risso et al, 2018)估计出的权重信息表现最好。不过仍需加权的普通转录组差异分析的独立比较研究进一步确认这个结果。
转录组差异分析工具的加权建模后效果的改善是以牺牲计算效率为代价的。随着单细胞数据中细胞量越来越大,算法的运行时间在方法选择中越来越重要。因此单细胞工具MAST被视作加权普通转录组差异分析工具的替代方法。MAST应用hurdle model消除dropout的影响,并构建基因表达相对于实验条件和技术协变量的变化模型。这是在前述评估研究中表现最好的单细胞差异基因分析方法 (Soneson & Robinson, 2018),并且在一个单一数据集的小范围研究中表现优于其它普通或单细胞差异基因分析方法 (Vieth et al, 2017)。MAST相比于加权的普通转录组差异分析工具有10-100倍的速度提升,还可以使用limma–voom模型进一步获得10倍的速度提升。虽然limma是普通转录组基因差异分析方法,但limma–voom与MAST性能相当。
因为用于差异基因分析的是标准化后但未进行技术变量和非相关生物变量校正的数据,因此在分析过程中考虑这些混杂变量对获得稳定的结果是关键的。通常差异基因分析工具都允许用户考虑混杂变量(confounders),但是把哪些变量纳入模型需要谨慎评估。比如,在多数单细胞数据集中,样本和实验处理条件是互斥的,因为几乎不可能对一个样品进行多个实验处理。如果我们在模型中同时考虑样品和实验条件两个协变量,跟这些协变量相关的变化则不会被明确确定。因此在考虑不同实验条件下的差异分析时,通常不在模型中引入样品信息作为协变量。当校正多个确定的批次分类变量时,可视化展示协变量的混杂影响将会变得更困难。在这个情况下,需要先确认模型设计矩阵是否满秩 (the model design matrix is full rank,full rank是指模型设计矩阵中的每一列都不能通过对其它列的线性组合得来,即它们之间不存在线性相关)。即便一些设计矩阵不是满秩,差异基因分析工具通常也不会给出警告而是继续运行。这时获得的结果将可能不是预期的分析方向。
我们这儿描述的场景中,实验条件协变量是在实验设计中决定的。因此在同一簇内基于这一协变量的差异基因分析是独立于聚类过程的。这将基于实验条件的差异基因分析和基于细胞簇的差异基因分析区别开来。基于实验条件的差异基因检测获得的P-values是显著性检验统计指标,需要进一步做多重检验校正。为了降低多重检验校正的压力,不感兴趣的基因通常需要预先排除掉。虽然假基因和非编码基因也可以提供有用信息,但在分析中也通常会被忽略掉。
陷阱和建议:
- DE测试不应基于校正后的数据(去噪,批次校正等),而应基于原始测量数据并在模型中引入干扰协变量。
- 用户不应依赖差异基因检测工具校正带有混杂协变量的模型。设计矩阵模型应当保证满秩。
- 我们建议使用
MAST或limma进行差异基因分析。
基因集分析。基因水平的分析通常会获得一长串难以解释其生物含义的基因列表。比如,处理组和对照样品通常会有数以千计差异表达基因。我们可以通过基因共有的特征对基因进行分组并检验这些特征在候补基因列表中的出现是否显著富集 (overrepresented)以便更好的解释结果。
不同用途的基因集信息可以在校正过的基因注释数据库中获取。为了解释差异基因结果,通常按共有的生物进程对基因进行分组。生物进程信息存储于MSigDB、Gene Ontology,通路数据存储于KEGG和Reactome数据库。基因列表的功能注释富集分析有多种工具可用,具体见Huang et al (2009)和 Tarca et al (2013)的综述。
分析领域富集分析的一个新操作是基于配对基因标记的配体-受体分析。通过受体和共轭配体之间的表达关系推断细胞簇之间的互作。CellPhoneDB提供了受体-配体对信息,并用统计模型解释簇之间的高表达基因是否可以配对 (Zepp et al, 2017; Zhou et al, 2017; Cohen et al, 2018; Vento‐Tormo et al, 2018)。
基因调控网络。基因不是独立发挥作用的。相反,基因的表达水平是由与其他基因和小分子之间的复杂调控决定的。揭示这些调控作用是基因调控网络(GRN)推断方法的目标。
基因调控网络推断是基于对基因共表达如相关性、互信息(mutual information)或回归模型分析进行测量(Chen&Mar,2018)。如果在考虑了其他所有基因都作为混杂因素后,两个目标基因之间依然存在共表达关系则可以推断这两个基因之间存在因果调控关系。基因调控关系鉴定与轨迹相关的调控基因检测相关。确实,几种单细胞GRN推论方法使用的机械微分方程模型中都引入了轨迹信息(Inferring gene regulatory relationships is related to the detection of trajectory‐associated regulatory genes. Indeed, several single‐cell GRN inference methods use trajectories with mechanistic differential equation models)(Ocone et al., 2015; Matsumoto et al., 2017)。
虽然存在专门针对scRNA-seq数据开发的GRN推断方法(SCONE:Matsumoto et al., 2017; PIDC:Chan et al., 2017; SCENIC:Aibar et al., 2017),但最近的比较分析表明普通和单细胞转录组的方法在这些数据表现都不好(Chen&Mar,2018)。GRN推断方法仍可提供识别生物过程的因果调节子的有用信息,但我们建议谨慎使用这些方法。
陷阱和建议:
- 用户应警惕推断的调控关系中的不确定性。基因调控关系丰富的基因模块比调控信息稀疏的模块更可靠。
分析平台。单细胞分析流程是多个独立开发的工具的集合。为了促进这些工具之间的数据传递,单细胞工具开发时都考虑使用一致的数据格式。这些工具为构建分析流程提供了基础。目前可用的分析平台有基于R(McCarthy et al., 2017; Butler et al., 2018)或Python(Wolf et al., 2018)的命令行,或具有图形用户界面(GUI)的本地应用程序(Patel,2018; Scholz et al., 2018)或Web服务(Gardeux et al., 2017; Zhu et al., 2017)。Zhu et al.(2017)和Zappia et al.(2018)对这些分析平台进行了综述。
在命令行平台中,Scater(McCarthy et al.,2017)和Seurat(Butler et al.,2018)可以轻松地与R Bioconductor项目提供的各种分析工具进行交互(Huber et al.,2015)。Scater在质量控制和数据预处理方面具有特殊优势,而Seurat可以说是最受欢迎和最全面的分析平台,提供了大量分析模块和教程。scanpy是一个基于Python的不断发展的新的单细胞分析平台,该平台可以扩展应用于分析更大规模的单细胞数据集。它可以整合Python写作的很多工具,尤其是在机器学习中流行的工具。
图形用户界面工具可以帮助非专业用户构建单细胞分析工作流。通常会基于固定的分析流程引导用户,这些分析流程有助于简化分析,但同时也限制了用户的灵活性,在探索性分析时特别有用。诸如Granatum(Zhu et al.,2017)和ASAP(Gardeux et al.,2017)之类的平台在集成的工具方面有所不同,其中Granatum包含了更多的方法。作为Web服务器,这两个平台都直接可用,但是其不同的计算框架将限制它们扩展到大规模数据集的能力。ASAP的测试数据集只包含92个细胞。基于Web的GUI平台的替代产品是FASTGenomics(Scholz et al., 2018)、iSEE(Rue-Albrecht et al., 2018)、IS-CellR(Patel,2018)和Granatum等软件包,它们在本地服务器上运行。这些平台和GUI封装工具可以随着本地电脑计算力的增强进行性能扩展。将来,Human Cell Atlas(https://www.humancellatlas.org/data-sharing)的不断发展将促进功能更强大的可应用于大规模细胞数据的可视化探索工具的开发。
结论与展望
我们综述了典型的scRNA‐seq分析流程的各个步骤,并在案例研究教程(https://www.github.com/theislab/single-cell-tutorial)中实现了这些步骤。本教程旨在通过对当前可用分析方法的比较研究确定当前最佳分析实践流程。虽然汇总单个最佳实践工具不能保证流程最佳,但我们希望我们的分析流程能够代表单细胞分析领域最新技术现状的一个掠影。它为新来者提供了进入该领域的合适切入点,并希望能有助于人类细胞图谱的scRNA-seq分析流程构建(Regev et al.,2018)。应该注意的是,方法比较必然落后于最新的方法开发。因此,我们提到了可能尚未被独立评估的新工具。随着新工具和更好工具的进一步发展和比较研究,文中推荐的每个单独的工具也需要一直更新,但是有关数据处理阶段的一般注意事项应保持不变 (生信宝典注:工具一直在变,但核心原理不变。来易生信,掌握核心知识。)。
特别受关注并且可能改变现有分析流程的两个开发方向是深度学习分析和单细胞多组学整合分析。由于深度学习可以很容易拓展到大规模数据分析,已经广泛用于计算机视觉到自然语言处理等多个领域,而且在基因组领域应用也越来越多。机器学习首先应用于scRNA-seq的降维和降噪分析领域,例如scVis:Ding et al.,2018; scGen:Preprint:Lotfollahi et al.,2018; DCA:Eraslan et al.,2019)。最近,深度学习已形成完善的分析流程用于拟合数据、对数据进行去噪并在模型框架内执行下游分析如聚类和差异表达等(scVI:Lopez et al., 2018)。在这一分析方法中,可以在保留数据变化的准确估计的同时,把噪声和批次效应的影响纳入下游统计分析模型中。诸如此类的整合建模方法有可能取代当前杂糅了多个工具的分析流程。
随着单细胞组学技术的进步,对组学分析流程开发的需求将会增长(Tanay&Regev,2017)。未来的单细胞分析平台将必须能够处理不同的数据源,例如DNA甲基化(Smallwood et al., 2014),染色质可及性(Buenrostro et al., 2015)或蛋白质丰度数据(Stoeckius et al., 2017),以及能对这些数据进行整合分析。如果是这个目的,将不能只使用我们流程起点的单个reads或计数矩阵作为输入。而且,整合RNA velocity分析的平台已经在适应多组学(多模态)数据结构,输入的是未剪接和剪接的reads数据(La Manno et al,2018)。单细胞多组学整合可以通过一致性聚类方法、多组学因子分析(Argelaguet et al., 2018)或多组学基因调控网络推断(Colomé-Tatché&Theis,2018)来实现。具有这些功能的分析流程将是未来开发的方向