

Hi-C分析需要的测序量比较高,1个样本往往需要测序很多的数据量,1个Hi-C文库可测序的数据量有限(一般情况下100-300G PE150,超出这个量,多测的数据可能含有较高的PCR dup),所以1个样本往往需要建几个文库来保证总的测序量足够。相同样本不同文库产生的数据需要有比较高的重复性,才能用于后续分析
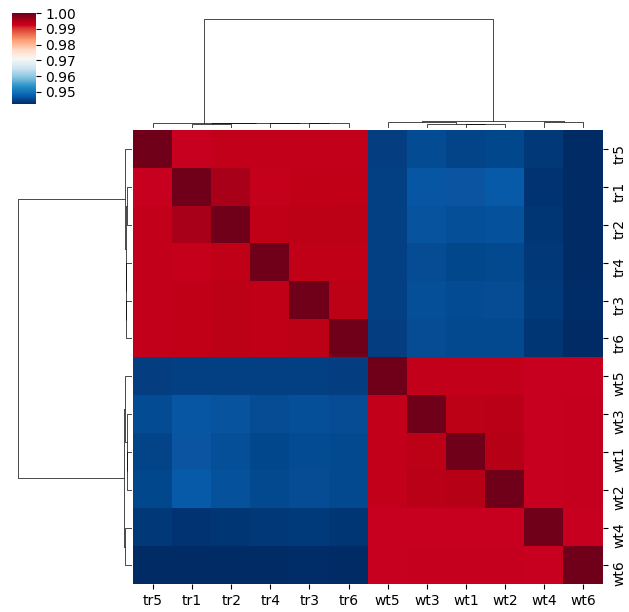
为了计算方便,开发了一个R脚本hicLibRepeatCor.R用于Hi-C文库相关性分析,计算两两Hi-C文库的cis矩阵相关性,对所有文库的相关性矩阵做聚类热图,用于衡量判断样本文库的重复性,和不同样本文库的差异
程序地址(hicLibRepeatCor.R)
- args <- commandArgs(TRUE)
- if (length(args) != 3){
- print("Rscript HicRepCorr.R ")
- print("Example : Rscript HicRepCorr.R raw.list 1000000 ./outdir")
- q()
- }
- library(HiTC)
- library(pheatmap)
- #args <- c("~/Desktop/hicrep/data/lib_matrix.list", 1000000, "./")
- panel.cor <- function(x, y, digits = 2, prefix = "", cex.cor, ...)
- {
- usr <- par("usr"); on.exit(par(usr))
- par(usr = c(0, 1, 0, 1))
- r <- abs(cor(x, y))
- txt <- format(c(r, 0.123456789), digits = digits)[1]
- txt <- paste0(prefix, txt)
- if(missing(cex.cor)) cex.cor <- 0.8/strwidth(txt)
- text(0.5, 0.5, txt, cex = cex.cor * r)
- }
- all_Intra_to_vector <- function(s1_hic, chrs){
- sample_ver <- c()
- for (chrom in chrs){
- s1_chr_mat <- log(as.vector(intdata(s1_hic[paste(chrom,chrom,sep="")][[1]])))
- s1_chr_mat[s1_chr_mat==-Inf]<-0
- sample_ver <- c(sample_ver, s1_chr_mat)
- }
- return(sample_ver)
- }
- raw_mats = read.table(args[1], header=F, stringsAsFactors=F)
- raw_mats = data.frame(subset(raw_mats, V2 == as.numeric(args[2])))
- intraHicVers <- list()
- bed_df <- read.table(raw_mats[1, "V4"])
- chrs <- unique(bed_df$V1)
- for (i in 1:nrow(raw_mats)){
- sample_intraHicObj <- importC(raw_mats[i,"V3"], raw_mats[i,"V4"],rm.trans=TRUE)
- #sample_intraHicObj <- sample_intraHicObj[isIntraChrom(sample_intraHicObj)]
- sample_intra_vers <- all_Intra_to_vector(sample_intraHicObj, chrs)
- intraHicVers[[raw_mats[i,"V1"]]] <- sample_intra_vers
- }
- intraHicVers <- do.call(cbind.data.frame, intraHicVers)
- cor_matr <- cor(intraHicVers)
- write.table(cor_matr, file.path(args[3], "./cor_matr.matrix"), sep="\t", quote=F, row.names=TRUE, col.names=TRUE)
- #pdf(NULL)
- pdf(file.path(args[3], "./pair.pdf"), w=7, h=7)
- pairs(intraHicVers, lower.panel = function(...) smoothScatter(..., add = T), upper.panel=panel.cor)
- dev.off()
- pdf(file.path(args[3], "./corr_heatmap.pdf"), w=7, h=7)
- par(font=2, font.axis=2,font.lab=2, mfrow=c(1,1))
- pheatmap(cor_matr, display_numbers = TRUE, number_format = "%.4f")
- dev.off()
程序用法
- Rscript hicLibRepeatCor.R <lib_matrix.list> <binsize> <outdir>
参数说明
lib_matrix.list
此参数为每一个文库的matrix列表,示例如下:
| wt1 | 1000000 | /abspath/wt1_1000000_symmetric.matrix | /abspath/wt1_1000000_abs.bed |
|---|---|---|---|
| wt2 | 1000000 | /abspath/wt2_1000000_symmetric.matrix | /abspath/wt2_1000000_abs.bed |
| tr1 | 1000000 | /abspath/tr1_1000000_symmetric.matrix | /abspath/tr1_1000000_abs.bed |
| tr2 | 1000000 | /abspath/tr2_1000000_symmetric.matrix | /abspath/tr2_1000000_abs.bed |
- 第一列:文库的命名
- 第二列:对应行矩阵的bin size
- 第三列:文库矩阵,格式同HiC-Pro软件buildMatrix产生的3列矩阵的格式
- 第四列:染色体位置与bin number对应关系bed文件,格式
HiC-Pro软件buildMatrix产生的bed文件
binsize
数字型,选择lib_matrix.list参数中第二列与binsize参数相同的matrix去做文库相关性分析
outdir
输出目录
输出结果示例
此程序会输出3个结果文件
- pair.pdf
- corr_heatmap.pdf
- cor_matr.matrix
pair.pdf
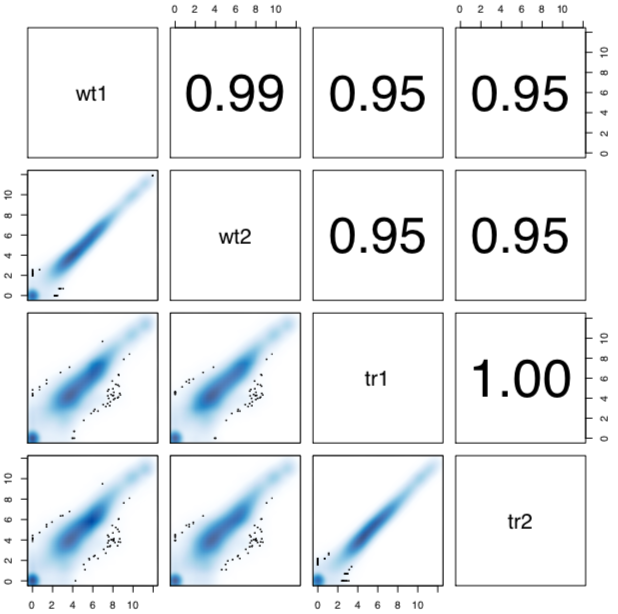

corr_heatmap.pdf
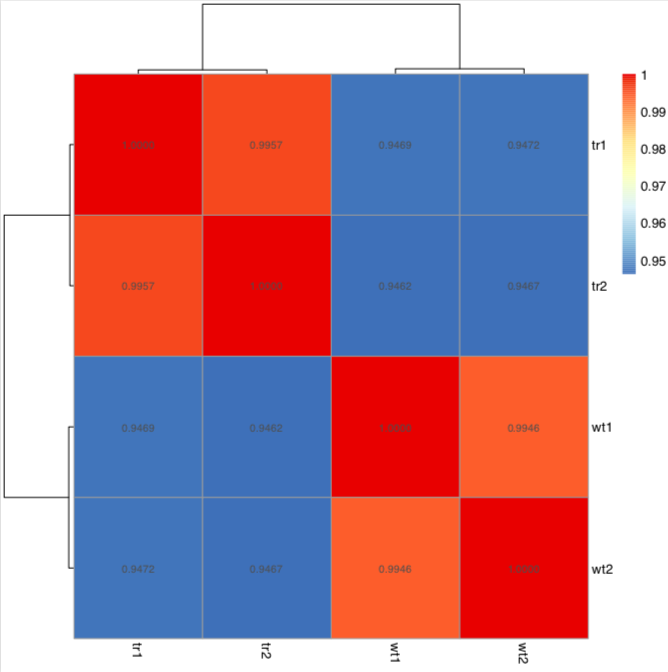
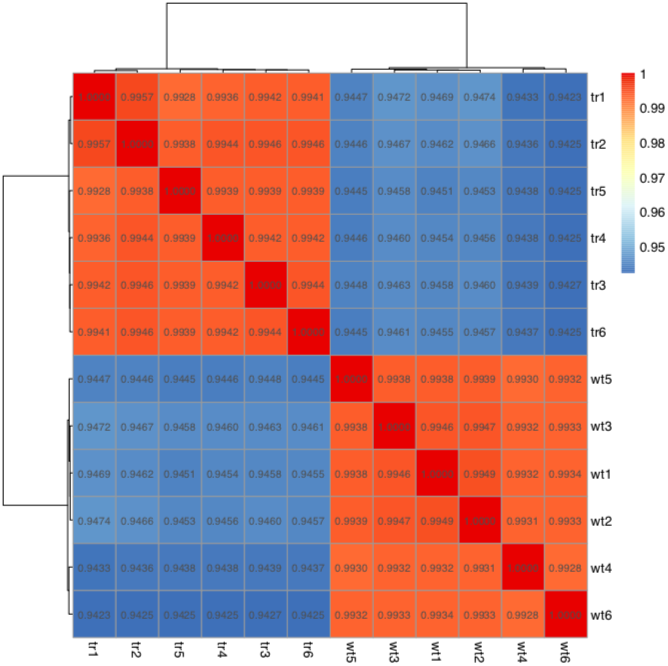
cor_matr.matrix
corr_heatmap.pdf的数据文件,对于有编程经验的用户可以自己再去根据自己的喜好去画热图
| wt1 | wt2 | tr1 | tr2 | |
|---|---|---|---|---|
| wt1 | 1 | 0.994581416401839 | 0.946872471200991 | 0.946223805271397 |
| wt2 | 0.994581416401839 | 1 | 0.947224994844614 | 0.946685529296875 |
| tr1 | 0.946872471200991 | 0.947224994844614 | 1 | 0.995740232735047 |
| tr2 | 0.946223805271397 | 0.946685529296875 | 0.995740232735047 | 1 |
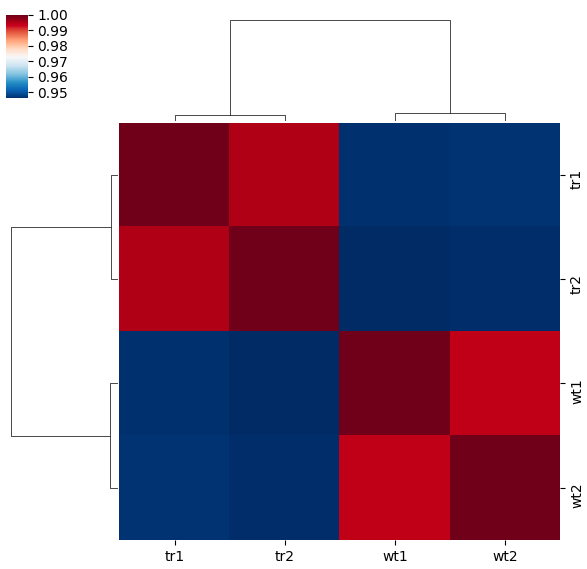
例如可以用以下python代码画图
- import pandas as pd
- import matplotlib
- import matplotlib as mpl
- # matplotlib.use('TkAgg') # for macbook
- # matplotlib.use('Agg') # linux or win
- import matplotlib.pyplot as plt
- import seaborn as sns
- df = pd.read_table("cor_matr.matrix", header=0, index_col=0 )
- sns.clustermap(df, cmap=sns.color_palette("RdBu_r", 7000))
- plt.show()
效果
程序依赖包
R环境需要安装以下两个R包才能使用此程序 * HiTC * pheatmap