

在上一期的《单细胞轨迹分析知多少--拟时间分析比较》中我们介绍了45种单细胞轨迹推断分析软件方法在以下4个方面的比较:
- 准确性
- 可扩展性
- 稳定性
- 可用性
得出了几项重要结论:
- 轨迹推断(TI,trajectory inference)软件方法输入和输出接口的标准化对于TI方法的广泛应用非常关键
- 不同TI方法的组合存在一定的互补性,能够高概率的得出与实际生物学模型相符的轨迹推断结果
- 在1个项目的数据集上尝试多种TI分析算法,一是可以提供多种算法证据支持,二是如果多种算法的结果有冲突有利于我们深度思考可能的具体生物学原因
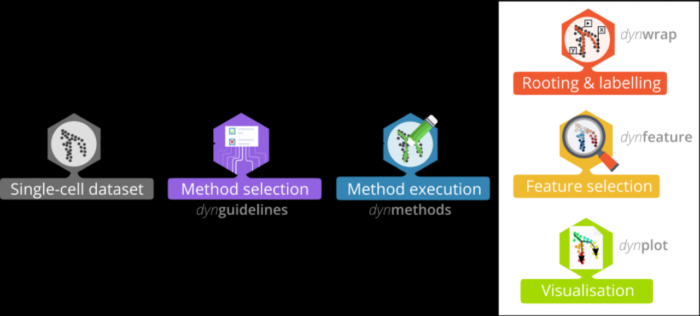
A comparison of single-cell trajectory inference methods(Saelens et al., 2019)的作者做了一个R包--dyno为终端用户提供完整的TI分析流程,dyno特点如下:
- 统一59种TI方法的输入输出接口
- 提供交互式指南工具,可帮助用户选择最合适的TI方法
- 简化了轨迹的解释和可视化,包括根据基因表达或者cluster着色
- 还可以进行下游分析,例如潜在marker gene 的鉴定
dyno安装
dyno是一个R包,需要R/Rstudio运行环境,目前的存放地址是在github- https://github.com/dynverse/dyno,所以我们用devtools进行安装,打开Rstudio,输入以下命令:
- #install.packages("devtools")
- devtools::install_github("dynverse/dyno")
在这个过程中会安装一系列dynverse R包(dynwrap, dynplot, dynmethods, …)。
如果是在linux系统使用,需要安装udunits和ImageMagick
- Debian / Ubuntu / Linux Mint:
sudo apt-get install libudunits2-dev imagemagick - Fedora / CentOS / RHEL:
sudo dnf install udunits2-devel ImageMagick-c -devel
运行TI方法需要用到Docker或者Singularity (version ≥ 3.0),如果电脑系统是Windows或者MacOS,推荐使用docker,如果是在集群,推荐使用 Singularity。
如果是win10系统,可以运行Docker CE。Singularity的安装请参考https://www.sylabs.io/docs/,如果已经安装了anaconda,可以执行conda create -n singularity -c conda-forge singularity=3.0.1安装
安装过程遇到的问题
API rate limit exceeded
因为dyno存放在了Github,在dyno的安装过程会使用GitHub API,默认的API限制是60次请求,所以会遇到API rate limit exceeded的问题,解决方式如下:
- 在Rstudio命令行执行
usethis::browse_github_pat()建议一个GitHub token - 执行
usethis::edit_r_environ()添加以下环境变量GITHUB_PAT = 'your_github_token - 重启R/Rstudio (重新载入 GITHUB_PAT)
- 再次重新安装:
devtools::install_github("dynverse/dyno")
其他安装问题
如果没有devtools是不能安装dyno的,所以有些时候devtools的安装也会出现一些问题,例如网络不稳定等,这时可以采用国内的源进行安装:install.packages('devtools', repos='https://mirrors.tuna.tsinghua.edu.cn/CRAN/')
在linux集群,如果没有root权限,安装使用dyno会非常困难,我准备了解决方案会在文章最后一节介绍。
windows10/MACOS用户快速体验方式
dyno的整个安装过程会非常漫长,如果是windows10或者MACOS用户可以在安装docker之后用我构建的R镜像:seqyuan/seqyuan-r:v0.0.1(R version 3.6.1)进行体验,这个R镜像安装了dyno、tidyverse及Seurat等包,可以免去安装过程快速体验dyno的强大,具体使用会在文章最后一节介绍。
dyno的使用
dyno的官网https://dynverse.org/有详细的使用步骤介绍,在这里我们就不再重复官网测试数据的结果,重点介绍Seurat分析的10X单细胞转录组数据的结果怎么样和dyno对接。
主要步骤
准备数据
为数据集选择最佳方法
运行TI方法
轨迹可视化
从生物学角度解释轨迹
- Rooting
- Milestone labelling
基因表达在轨迹上的展示
- A global overview of the most predictive genes
- Lineage/branch markers
- Genes important at bifurcation points
准备数据
首先打开Rstudio(如果是linux,打开R命令行),载入以下R包
- library(dyno)
- library(tidyverse)
- library(Matrix)
- library(Seurat)
dyno对数据的包装主要通过dynwrap包来实现
Gene expression data
dynwrap要求raw counts和normalised(log2)表达数据,低表达的细胞、双细胞、坏细胞应该被提前过滤。使用Seurat处理后的数据一般都包含了这些步骤。
wrap_expression要求raw counts和normalised表达为 sparse matrix (dgCMatrix)(列为genes/features,行为细胞)
- #读入seurat处理后的rds文件
- sdata <- readRDS(file = "/Users/yuanzan/Desktop/tmp/seurat_project.rds")
- #添加raw counts和normalised expression
- #seurat的矩阵需要进行行列转换,以使行为细胞,列为基因
- dataset <- wrap_expression(
- counts = t(sdata@assays$RNA@counts),
- expression = t(sdata@assays$RNA@data)
- )
- #添加先验信息,这里添加的是开始转换的“细胞id”,后期可视化可以根据具体的轨迹推断结果进行调整
- dataset <- add_prior_information(
- dataset,
- start_id = "TTTGTTGCAACTCATG.wt"
- )
- #添加数据的cluster信息,这里我们直接用“seurat_clusters”即可
- dataset <- add_grouping(
- dataset,
- sdata$seurat_clusters
- )
为数据集选择最佳方法
如果是MACOS,可以执行以下命令以获取推荐的最佳TI方法
- guidelines <- guidelines_shiny(dataset)
- methods_selected <- guidelines$methods_selected
执行guidelines <- guidelines_shiny(dataset)命令之后会弹出一个shiny页面,如下图
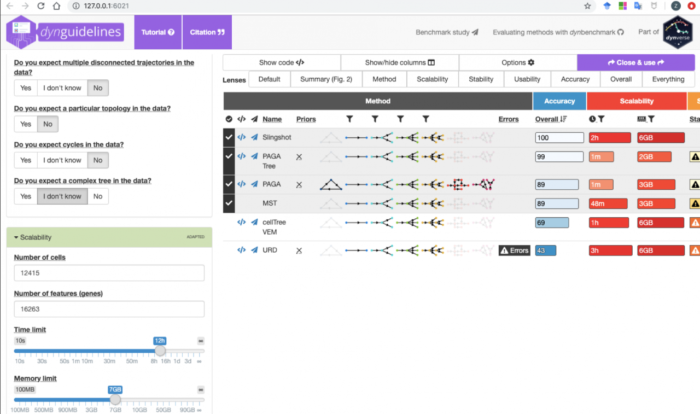
因为我们给了dataset,所以guidelines页面会自动填写细胞数和基因数参数,我们再把预期的运行时间和运行内存选择设置一下,可以扩大选择TI方法的选择范围。
通过选择对于推断轨迹的预期以及左侧关于内存、运行时间等各项参数,guidelines为我推荐了几个TI算法的组合,点击右上角的Close & use关闭这个shiny页面。

methods_selected <- guidelines$methods_selected这个命令是把guidelines推荐给我们的方法赋值给methods_selected,这是一个字符串向量。

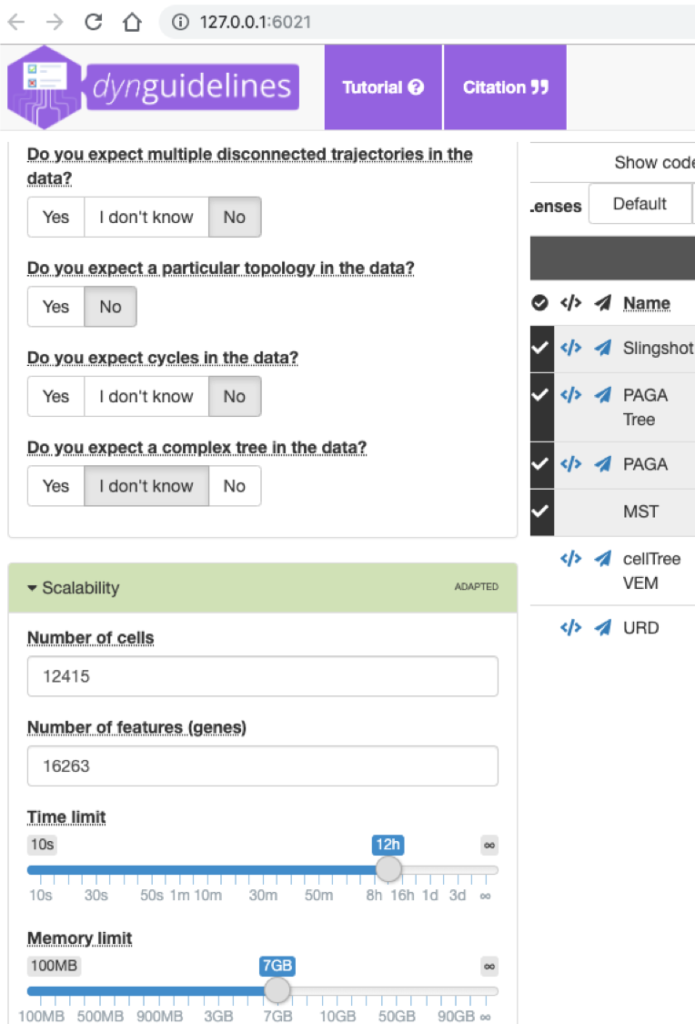
如果我们是在linux集群上进行的操作,这一步弹出guidelines shiny的界面可能需要通过很复杂的设置才能获得,而且也不利于把命令行写成脚本执行。幸好我们有替代方法,那就是
- 通过命令
dim(dataset$counts)获取cell number和feature/gene number - 打开网址
http://guidelines.dynverse.org - 在guidelines网页填写细胞数、gene数以及其他设置
- 打对勾的TI方法就是推荐给我们的,如下图

运行TI方法
- model_slingshot <- infer_trajectory(dataset, methods_selected[1])
- model_paga_tree <- infer_trajectory(dataset, methods_selected[2])
- model_paga <- infer_trajectory(dataset, methods_selected[3])
- model_mst <- infer_trajectory(dataset, methods_selected[4])
作者把相应TI方法的源码封装在了docker镜像里,调用相应方法的后台操作其实是:先拉取TI方法对应的镜像,然后再R进程内部启动docker容器执行算法,所以如果你使用的方法事先没有经过
docker pull,那么算法的执行时间其实包含了拉取镜像所需要的时间,建议把常用到的TI方法先pull到本地,这样就能使脚本代码的执行更快。
目前作者封装的59种TI方法列表见网址:https://github.com/dynverse/dynmethods#list-of-included-methods,用到代码中的调用一般是单词的所有字母都小写,单词之间加下划线。
guidelines推荐给我的methods_selected包含4个TI方法,本文开头介绍了《单细胞轨迹分析知多少--拟时间分析比较》得出了几项重要结论,理论指导实践,所以我打算把4中TI方法都试一下,看看结果怎么样。
简单可视化
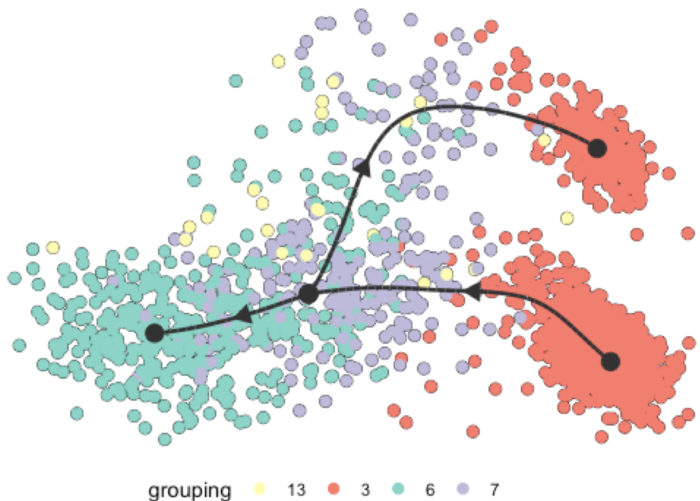
- model <- model_slingshot
- plot_dimred(
- model,
- expression_source = dataset$expression,
- grouping = dataset$grouping
- )
slingshot
paga
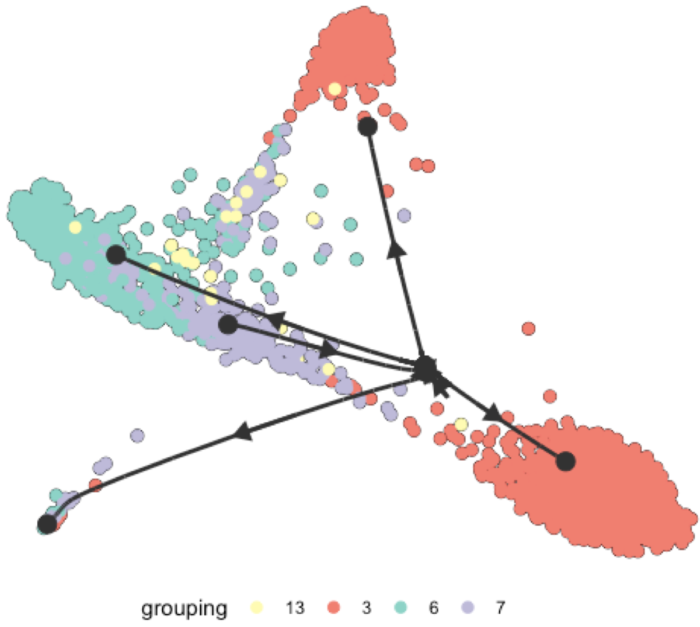
mst

paga_tree

从生物学角度解释轨迹
Rooting
大多数方法没有直接的方法来推断轨迹的方向性。在这种情况下,应该使用一些外部信息来“确定”轨迹,例如使用一组marker基因
添加rooting gene列表,这里需要根据具体的生物学问题来定,为了更快速的演示,我不再深究,这里仅给一个示例作为演示用
- # 初步来看我认为slingshot方法得到的结果和我的预期比较相近,后续将以此方法得到的model进行后续演示
- model <- model_slingshot
- model <- model %>%
- add_root_using_expression(c("KLRB1"), dataset$expression)
Milestone labelling
可以使用marker基因标记里程碑,这些标签随后可用于后续分析和可视化
- # 通过model$milestone_ids我们知道了有4个里程碑
- # 这个示例我们标记3个里程碑,此处仅为示例,请忽略标签意义
- model <- label_milestones_markers(
- model,
- markers = list(
- Tn = c("CCR7"),
- NK = c("KLRD1"),
- CTL = c("GZMK")
- ),
- dataset$expression
- )
轨迹可视化
dyno提供了降维、轨迹模型和细胞聚类等可视化展示方法
细胞亚群在轨迹上的展示
- model <- model %>% add_dimred(dyndimred::dimred_mds, expression_source = dataset$expression)
- plot_dimred(
- model,
- expression_source = dataset$expression,
- grouping = dataset$grouping,
- label_milestones = TRUE
- )
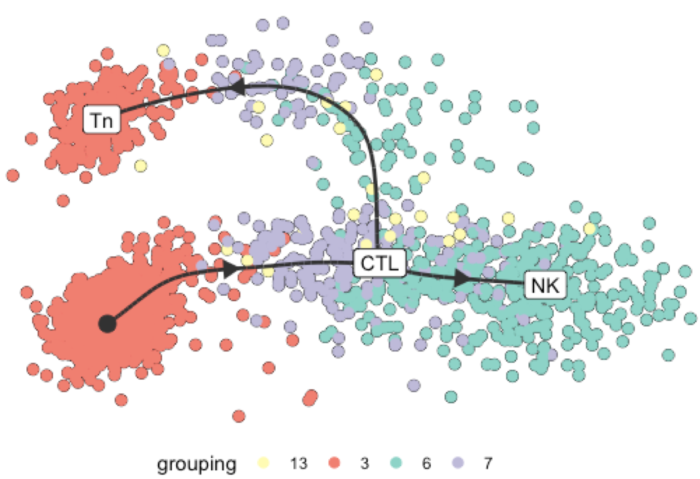
通过基础的4个细胞亚群的轨迹分布,我们明显的看到3亚群分为了2部分,所以我打算后续调整第3群为2个亚群再做轨迹分析,以便更好的体现它们的差异。
我们设置了label_milestones = TRUE,所以里程碑被标注到了轨迹模型上,model一共有4个里程碑我们只标注了3个所以上图只显示3个。

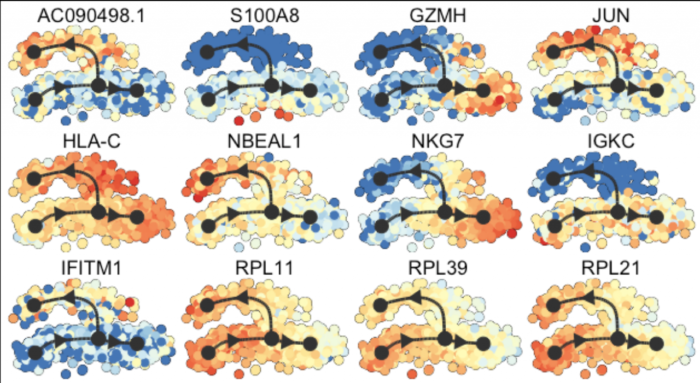
基因表达在轨迹上的展示
这里我们做CCR7和CCL5两个基因表达示例
- plot_dimred(
- model,
- expression_source = dataset$expression,
- color_cells = "feature",
- feature_oi = "CCR7",
- color_density = "grouping",
- grouping = dataset$grouping,
- label_milestones = TRUE
- )
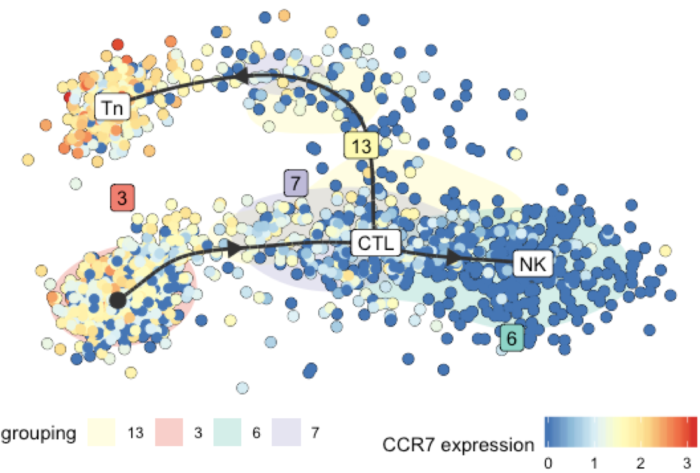
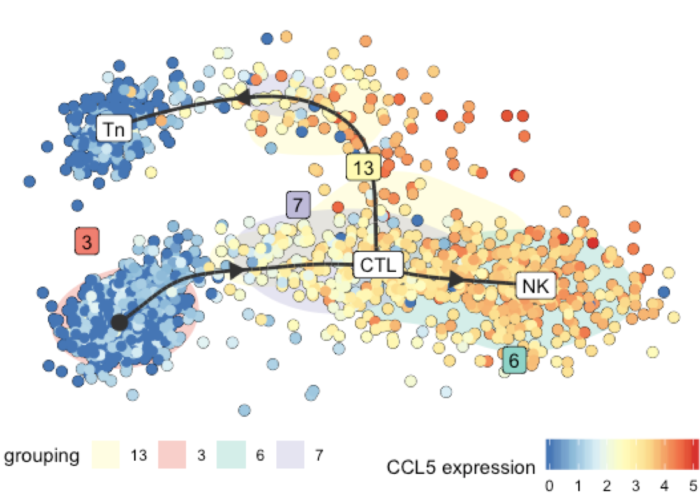
在展示基因表达的同时,不同细胞亚群以细胞的背景颜色表示,以下官方示例展示的更清楚一些
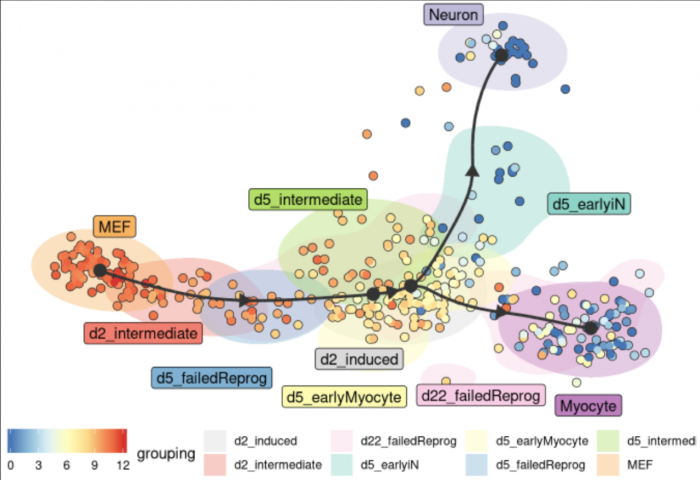
预测和可视化感兴趣的基因
dyno整合了几种方法来从轨迹中提取候选标记基因/特征
最具预测性的基因的全球概述
默认情况下,绘制热图时会计算总体上最重要的基因
- plot_heatmap(
- model,
- expression_source = dataset$expression,
- grouping = dataset$grouping,
- features_oi = 50
- )
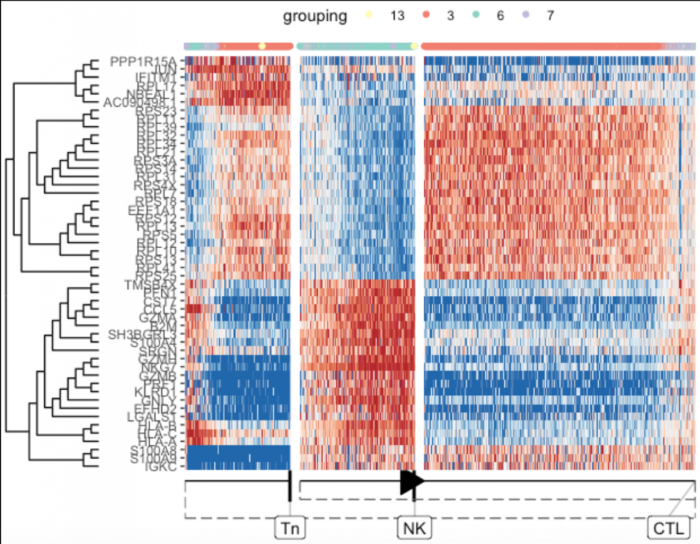
Lineage/branch markers
dyno还有提取特定分支的功能,例如:当细胞分化成神经元时改变的基因
- branch_feature_importance <- calculate_branch_feature_importance(model, expression_source=dataset$expression)
- select_features <- branch_feature_importance %>%
- filter(to == which(model$milestone_labelling =="CTL")) %>%
- top_n(30, importance) %>%
- pull(feature_id)
- plot_heatmap(
- model,
- expression_source = dataset$expression,
- features_oi = select_features
- )
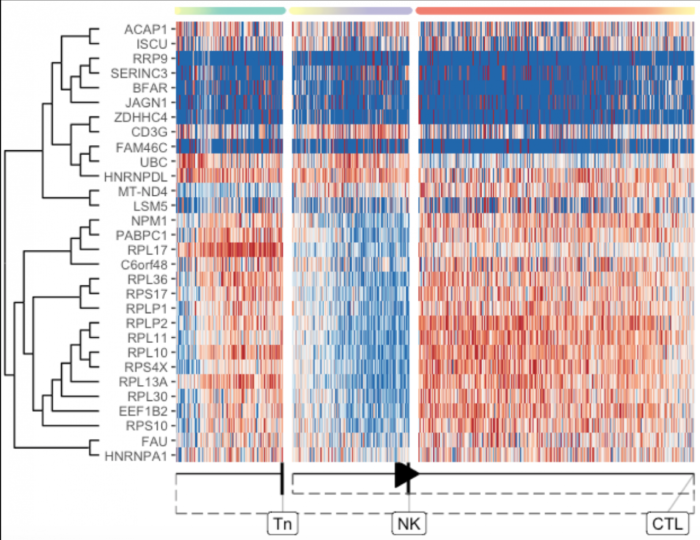
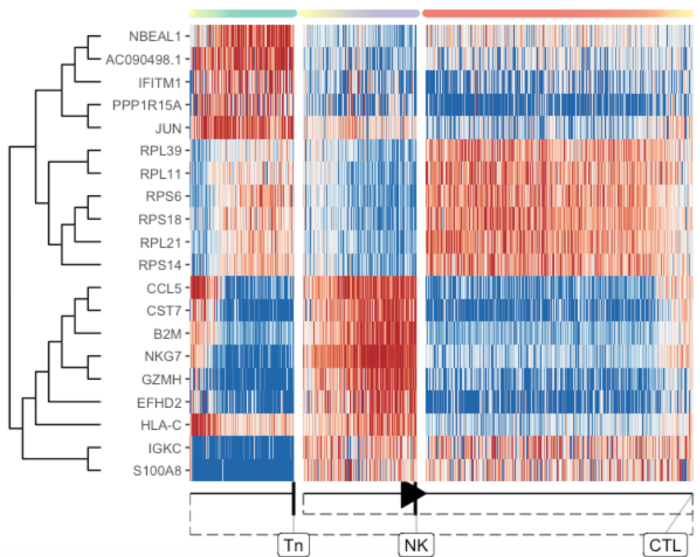
分叉点重要的基因
提取在分支点发生变化的特征/基因
- branching_milestone <- model$milestone_network %>% group_by(from) %>% filter(n() > 1) %>% pull(from) %>% first()
- branch_feature_importance <- calculate_branching_point_feature_importance(model, expression_source=dataset$expression, milestones_oi = branching_milestone)
- branching_point_features <- branch_feature_importance %>% top_n(20, importance) %>% pull(feature_id)
- plot_heatmap(
- model,
- expression_source = dataset$expression,
- features_oi = branching_point_features
- )
- space <- dyndimred::dimred_mds(dataset$expression)
- map(branching_point_features[1:12], function(feature_oi) {
- plot_dimred(model, dimred = space, expression_source = dataset$expression, feature_oi = feature_oi, label_milestones = FALSE)
- theme(legend.position = "none")
- ggtitle(feature_oi)
- }) %>% patchwork::wrap_plots()
快速免安装体验方式
对于MACOS或者有root权限的用户,如果想快速体验dyno,可以在安装并启动docker之后,执行以下命令行进入docker环境命令行,然后打开R终端。
- sudo docker run -it --memory=20G --memory-swap=20G --oom-kill-disable -v /home/zanyuan:/zanyuan -v /tmp:/tmp -v /usr/bin/:/dockerbin -v /var/run/docker.sock:/var/run/docker.sock seqyuan/seqyuan-r:v0.0.1 /bin/bash
理解上面的代码可能需要一些docker方面的知识,其中-v /home/zanyuan:/zanyuan 这一行指的是本地的存储挂载为docker容器里的/zanyuan地址;-v /tmp:/tmp是临时存储目录挂载;-v /usr/bin/:/dockerbin指的是docker所在本地路径挂载为容器里的/dockerbin;seqyuan/seqyuan-r:v0.0.1这个是docker启动的镜像名称,我构建的dockerfile请见docker hub官网。
如果运行时间比较长,可以把代码写到一个名为test_dyno.R脚本里例如:
- library(dyno)
- library(tidyverse)
- library(Matrix)
- library(Seurat)
- pso <- readRDS(file = "/zanyuan/data/cd8_t1.rds")
- dataset <- wrap_expression(
- counts = t(pso@assays$RNA@counts),
- expression = t(pso@assays$RNA@data)
- )
- dataset <- add_prior_information(
- dataset,
- start_id = "AGCGTCGTCCCTAACC.5"
- )
- model <- infer_trajectory(dataset, "paga")
- saveRDS(model, file = "/zanyuan/data/cd8_paga.model", ascii = FALSE, version = NULL, compress = TRUE, refhook = NULL)
然后执行
- sudo docker run -it --memory=20G --memory-swap=20G --oom-kill-disable -v /home/zanyuan:/zanyuan -v /tmp:/tmp -v /usr/bin/:/dockerbin -v /var/run/docker.sock:/var/run/docker.sock seqyuan/seqyuan-r:v0.0.1 Rscript /zanyuan/test_ti.R
后续
后续会推出在linux集群无root权限的情况下调用singularity来执行dyno