

由于最近的课题需要使用单细胞数据,因此开始学习单细胞的一系列分析方法和流程,这里就记录一下使用seurat进行单细胞RNA-seq聚类分析的流程,包括一些降维的知识,函数的调用等等。降维算是一件不大不小的事情,你说他简单吧,好像也没那么简单,你说他到底有多重要吧,似乎也给不出什么明确的结论,更多的是验证一下数据的可靠性,为后面的研究提供一些猜想等等。所以吧,降维这事,不讲不行,但讲得太深也没必要(目前这么觉得,可能随着研究继续深入就真香了…),所以这一期就不从最基础的PCA开始说了,而是简单记录一下tSNE的一些基础和想法,更详细的关于降维的算法和理解可能会在后面几期中再做。OK,那么我们结合代码直接开始。
这里的教程是直接根据seurat官网的guide来做的,数据也是官网提供的测试数据,点击这里下载后解压到工作文件夹。
- setwd('your/path/to/seurat/filtered_gene_bc_matrices/hg19')
- #载入所需要的包
- library(dplyr)
- library(Seurat)
- #读入数据,创建seurat对象
- pbmc.data <- Read10X('./')
- pbmc <- CreateSeuratObject(counts = pbmc.data,project = 'pbmc3k',min.cells = 3,min.features = 200)
直接用Read10X读入原始数据,如果直接给了UMI matrix就直接read.table就行。在创建seurat对象过程中会先进行一步初筛,筛去包含细胞太少的feature(这里是3),以及包含feature(也就是gene)太少的细胞(这里是200),这些数据基本都是无效数据。project参数基本就是个命名,3k的意思是大约有3000个细胞,其实是2700个。
- #简单看一下seurat对象的数据结构,count matrix每一列为一个细胞,每一行为一个feature(基因)
- #这样一个count matrix存储在seurat[['RNA']]@count中,可以对应地去看一下
- pbmc.data[c("CD3D", "TCL1A", "MS4A1"), 1:30]
- pbmc[['RNA']]@counts[c("CD3D", "TCL1A", "MS4A1"),1:30]
这两者的返回值应该是完全对应的,目的就是提示自己注意一下数据结构。
- > pbmc.data[c("CD3D", "TCL1A", "MS4A1"), 1:30]
- 3 x 30 sparse Matrix of class "dgCMatrix"
- [[ suppressing 30 column names ‘AAACATACAACCAC’, ‘AAACATTGAGCTAC’, ‘AAACATTGATCAGC’ ... ]]
- CD3D 4 . 10 . . 1 2 3 1 . . 2 7 1 . . 1 3 . 2 3 . . . . . 3 4 1 5
- TCL1A . . . . . . . . 1 . . . . . . . . . . . . 1 . . . . . . . .
- MS4A1 . 6 . . . . . . 1 1 1 . . . . . . . . . 36 1 2 . . 2 . . . .
- > pbmc[['RNA']]@counts[c("CD3D", "TCL1A", "MS4A1"),1:30]
- 3 x 30 sparse Matrix of class "dgCMatrix"
- [[ suppressing 30 column names ‘AAACATACAACCAC’, ‘AAACATTGAGCTAC’, ‘AAACATTGATCAGC’ ... ]]
- CD3D 4 . 10 . . 1 2 3 1 . . 2 7 1 . . 1 3 . 2 3 . . . . . 3 4 1 5
- TCL1A . . . . . . . . 1 . . . . . . . . . . . . 1 . . . . . . . .
- MS4A1 . 6 . . . . . . 1 1 1 . . . . . . . . . 36 1 2 . . 2 . . . .
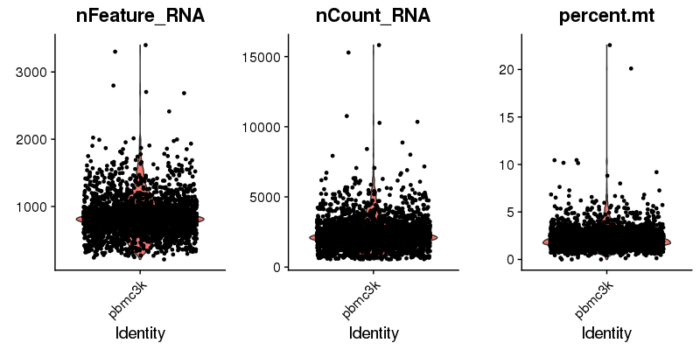
读取完数据后,思路也是挺一般的,一定是先质控,然后标准化归一化,接着降维聚类,最后寻找marker基因。质控包括质控线粒体基因数以及单个细胞所检测到的所有基因数。在衰老损伤或凋亡的细胞中,往往能检测出极端的线粒体基因数,这类细胞大部分情况下都不是我们想要的‘正常’细胞,除非你专门研究衰老凋亡。另外呢,如果一个细胞检测到的基因数太少,那这个细胞的质量大概率有问题,如果检测到的基因太多,那这个细胞很有可能是几个细胞粘在一起未分开的细胞团。
- #线粒体基因质控
- pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
- VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
- #质控reads数和基因数的关系
- plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
- plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
- CombinePlots(plots = list(plot1, plot2))
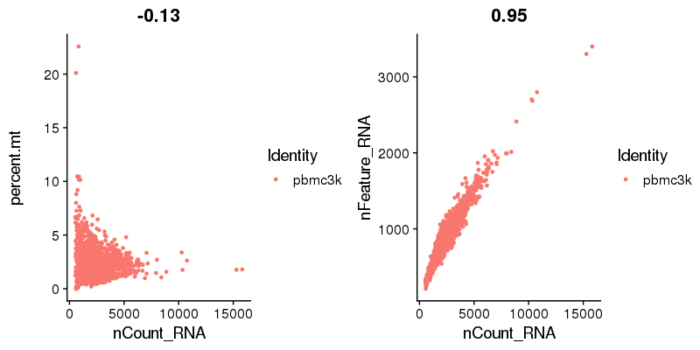
从这两张图上来看,基因数超过2500基本上就是outlier,线粒体基因数则卡到5%。
- #挑选基因数大于200小于2500,线粒体基因数小于5%的数据(data-selection)
- pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
- #再来看下小提琴图
- VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
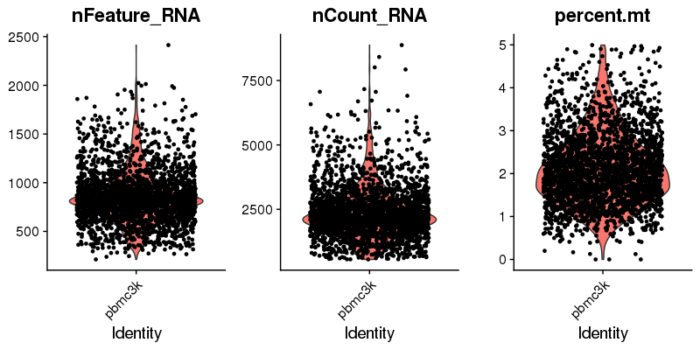
和前面的小提琴图相比,明显可以看到outlier被去除。筛选完数据后进行标准化。
- #筛选完数据,进行标准化
- pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
- #这个scale factor就是默认值可以不改,也可以不填这个参数
单细胞数据有一个很大的特点是数据量大,单个细胞数据的噪声高,为了优化后续PCA降维的计算,因此要从中选择最差异表达的基因进行后续操作。如果不这么做,那么PCA面临的是13714维的数据,并且包含了2700个点(这已经是筛选过的测试数据集了)。所以这里先挑选了2000个在细胞与细胞之间表达差异最大(方差)的基因,用于后续分析,当然你也可以适当调整基因数。
- #寻找差异表达基因(细胞与细胞之间方差大)这也很好理解,基因的个数可以根据数据和后续分析进行调整
- pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
- #来看下这些差异表达的基因
- #数据存储在seurat[['RNA']]@var.features,可以用VariableFeatures函数进行调用
- top10 <- head(VariableFeatures(pbmc), 10)
- plot1 <- VariableFeaturePlot(pbmc)
- plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
- CombinePlots(plots = list(plot1, plot2))
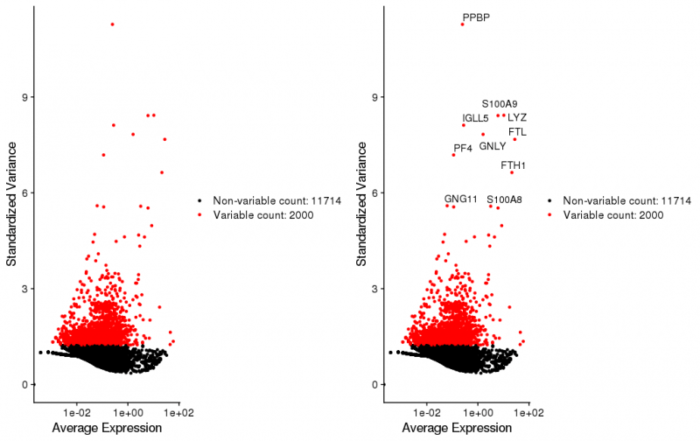
最后再对所有基因进行scale,将均值中心化为0,将方差齐至1,防止部分feature由于本身值比较大从而主导了差异。这一步的计算会很慢,因此也可以只scale我们上面选出来的那2000个基因,只要将ScaleData中的features参数删去,就会默认只scale这2000个基因。
- #scale,feature均值中心化为0,方差为1的归一化操作,降低一些高表达feature的主导程度
- #数据存储在seurat[['RNA']]@scale.data
- all.genes <- rownames(pbmc)
- pbmc <- ScaleData(pbmc, features = all.genes)
处理完数据就可以进行聚类了。由于上文提到,单细胞RNA-seq的噪声大,数据量大,因此直接用基因进行降维和聚类往往会引入大量噪声和无用的计算,因此这里采用了先进行PCA线性降维,将所有的信息合并后进行处理,从而降低噪声所占的比例或者说是主导程度,再进行聚类的方法。OK,那么我们先来看PCA降维。
- #PCA降维,高维向量是所选择的2000个高差异表达基因
- pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
- print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
输出结果:
- > print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
- PC_ 1
- Positive: CST3, TYROBP, LST1, AIF1, FTL
- Negative: MALAT1, LTB, IL32, IL7R, CD2
- PC_ 2
- Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1
- Negative: NKG7, PRF1, CST7, GZMB, GZMA
- PC_ 3
- Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1
- Negative: PPBP, PF4, SDPR, SPARC, GNG11
- PC_ 4
- Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1
- Negative: VIM, IL7R, S100A6, IL32, S100A8
- PC_ 5
- Positive: GZMB, NKG7, S100A8, FGFBP2, GNLY
- Negative: LTB, IL7R, CKB, VIM, MS4A7
将结果可视化,画出PCA降维图,以及PC轴中基因的贡献。
- #可视化
- VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
- DimPlot(pbmc, reduction = "pca")
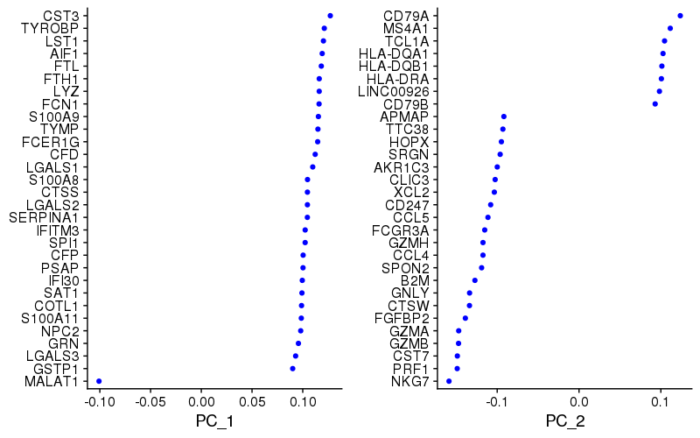
这张图表示了PC1和2的构成情况,也可以理解成不同基因对PC轴的贡献。画出这张图,包括下面的不同PC轴的热图,其目的都是为了将基因和降维关联起来,如果在某条PC轴上看到了与所研究的生物学问题相关的一些marker基因,那就能提示我们这条PC轴可能能将我们想要的一群细胞区分开。
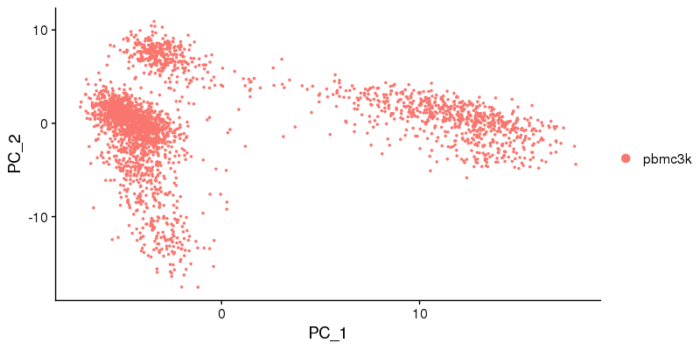
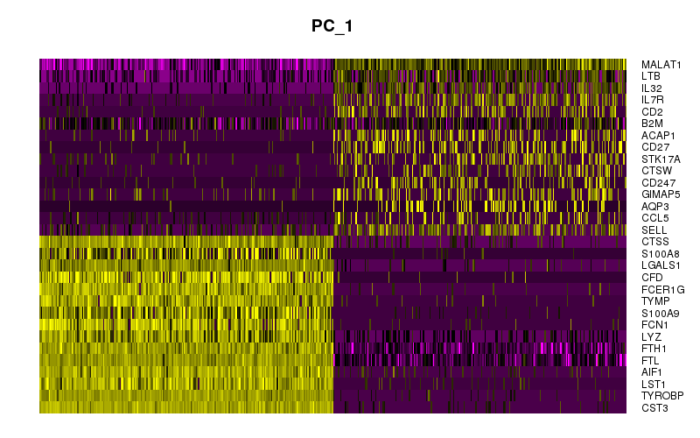
同样的,我们可以用热图的形式更好的去观察异质性的来源,从而将基因和降维联系起来。
- #用heatmap查看每一个PC上异质性的来源,横轴是选择的最‘极端’的细胞
- DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)
- DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)
当然也可以画出其他PC的热图,由于图片太大太长,这里就不展示了。OK,做完PCA后我们就可以考虑聚类了。那么还是回到上面的问题,单细胞RNA-seq数据量大,噪音也大,我要选择哪些轴作为我进一步聚类的依据呢?这里就要对不同的主成分的显著性或贡献值进行比较了。这里介绍两种方法。
JackStraw法,是随机地从所有的feature中选出1%,去计算PCA值,再将这部分基因的PCA值与实际观察到的PCA值进行比较,从而得到统计显著性,最终的结果是将每个基因与不同主成分的相关性的显著性水平计算出来。那么一个PC越显著,对结果的影响越大,那么它就应当更多的富集那些显著的feature(基因)。更多的原理参考文献Macosko et al。
- #单细胞RNA-seq的噪声大,因此采用PCA值作为聚类的依据,那么如何判断采用哪些PC或多少PC?
- #JackStraw法
- pbmc <- JackStraw(pbmc, num.replicate = 100)
- pbmc <- ScoreJackStraw(pbmc, dims = 1:20)
- JackStrawPlot(pbmc, dims = 1:15)
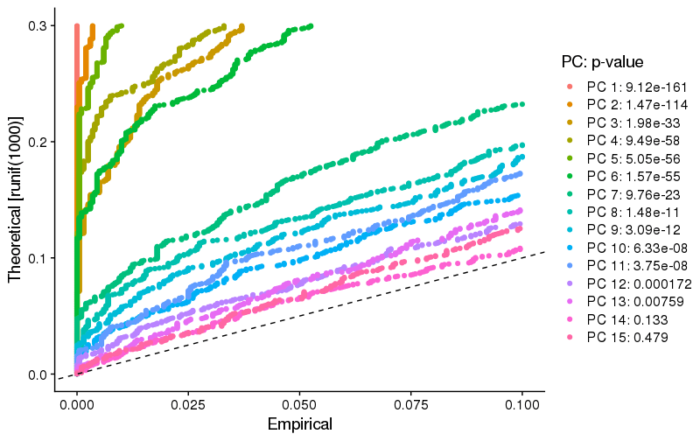
这张图虚线表示null distribution,即PC的显著性越强,则包含的显著性高的feature就越多,因此如果一个PC显著,则它在这张图上将显著地左偏。因此我们认为前10-12个PC较为显著,可以作为我们后续聚类的依据。需要注意的是,对于较大的数据量,JackStraw法的计算速度相当慢。
另一种方法是去看每个PC轴对变异的贡献值情况,也称弯道图(Elbow plot)法。
- #elbowplot
- ElbowPlot(pbmc)
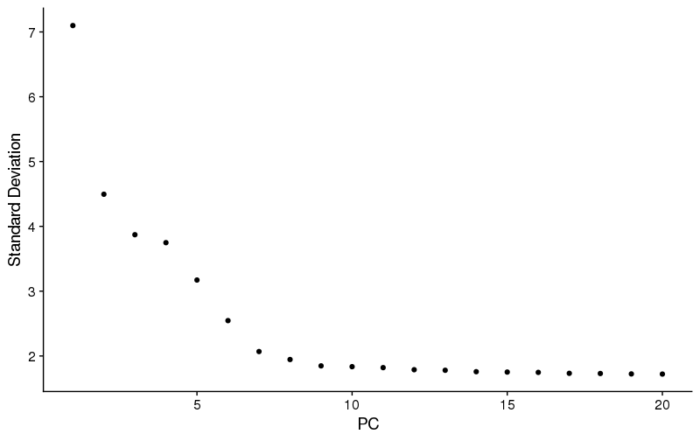
可以看到PC的贡献值不断减小,到PC10后趋于稳定(噪声主导),因此最终我们选择前10个PC作为我们后面聚类的依据。这里再说下选择PC的两个注意点,一是最显著的PC不一定能帮助我们区分一些很小的类,这些类由于本身数量较少,因此很容易淹没在噪声中;二是如果对取多少PC或哪些PC没有把握,则尽量往多了取,噪声虽然会更多,但总比信息丢失要好。OK,选择好了PC作为新的高维空间(没那么高,也就10维),就可以去进行聚类了。
Seurat所用的是基于流形学习的KNN算法。流形学习假设数据是均匀采样于一个高维欧氏空间中的低维流形,流形学习就是从高维采样数据中恢复低维流形结构,即找到高维空间中的低维流形,并求出相应的嵌入映射,以实现维数约简或者数据可视化。它是从观测到的现象中去寻找事物的本质,找到产生数据的内在规律。我们可以想象一下,假设我们的三维空间中有一些点,这些点分布在三维空间的一张曲面上(高维空间中的低维流形),我们要做的就是去描述这些点在这张曲面上的排列规律。自然我们要通过距离去描述这样的排列规律,但曲面上的距离和空间中的距离不同,这里再打个比方。想象一下地球这个曲面,一个人在北极,一个人在南极,空间上的距离是地球的直径,但在曲面上的距离,则是和经线重合的一个半圆。因此我们并不能直接用空间距离去刻画曲面上点与点之间的位置关系。但这并不是绝对不行,或者说是通过近似可行,这怎么理解呢。继续用地球类比,一个人在北京四环,一个人在北京五环,他们之间的距离需要用曲面这么复杂的图形来度量吗?不需要吧,可以直接近似成平面吧!这就引出了KNN图。KNN算法,即K近邻算法是一种监督学习算法,本质上是要在给定的训练样本中找到与某一个测试样本A最近的K个实例,然后统计k个实例中所属类别计数最多的那个类,就是A的类别。这怎么理解呢,就是把曲面上临近的点连成空间中的直线,用折线去近似曲面上的距离,说的再简单一点就是把地球上所有邻近的人都用直线连起来,然后去寻找把北极的人和南极的人连起来的最短折线,那么这条折线的距离就近似于南极的人到北极的人的曲面上的距离。构建好KNN图,定义好流形上不同点之间的距离,就可以根据这个距离进行聚类了,在曲面上距离近的点就会被聚成一类。
- #最终选择前10个pc作为聚类的依据
- #resolution的参数可以具体调节
- pbmc <- FindNeighbors(pbmc, dims = 1:10)
- pbmc <- FindClusters(pbmc, resolution = 0.5)
其中resolution的参数可以自己调节,resolution越大,所聚出的类的数目也越多,一般来说对于3k个细胞,推荐值为0.4-1.2,可根据自己的数据和先验知识或直觉进行调整。
可以查看前5个细胞的聚类情况。
- > head(Idents(pbmc), 5)
- AAACATACAACCAC AAACATTGAGCTAC AAACATTGATCAGC AAACCGTGCTTCCG AAACCGTGTATGCG
- 0 3 2 1 6
- Levels: 0 1 2 3 4 5 6 7 8
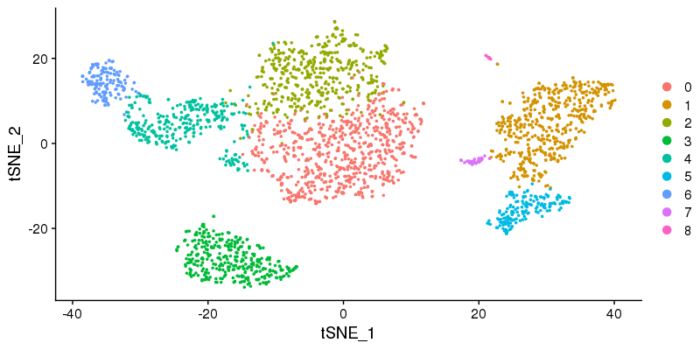
总共聚出9类,9个level。聚完类就要对聚类进行可视化了,可以采用tSNE或UMAP来进行非线性降维。这里简单提一下tSNE,tSNE是从SNE发展而来,SNE的想法是说高维空间中相似的点(近),在低维空间中也是相似的(近),当然一般会考虑用欧式距离直接来表征,但SNE则将这种距离关系转换为一种条件概率来计算,如果i和j两个点临近,则i周围出现j的条件概率就大,无论在高维空间,还是低维空间。SNE选择采用高斯分布来衡量这种条件概率。当然SNE的想法非常好,但效果却不是特别好,在SNE的基础上发展起来的tSNE则显著提高了降维可视化的效果,tSNE的想法是在高维空间用高斯分布去描述条件概率,在低维空间则用t分布去描述,这样的好处是t分布比标准正态更‘胖’,更长尾,因此在低维空间使用t分布,可以使数据之间分散的更开,降维可视化的效果便得到了提升。
- #非线性降维UMAP或tSNE
- pbmc <- RunUMAP(pbmc, dims = 1:10)
- DimPlot(pbmc, reduction = "umap")
- pbmc <- RunTSNE(pbmc, dims = 1:10)
- DimPlot(pbmc, reduction = "tsne")
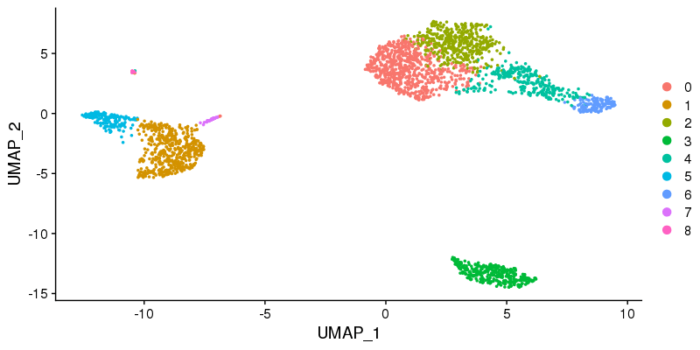
UMAP图,tSNE图可能会样子有所差异,但本身其坐标没有直接的物理意义,因此我们更关心的是聚类的情况,以及类与类之间的关系。
最后就是寻找差异表达基因,然后identify各个cluster了。当然这里需要注意的是,默认情况下,既会有正marker,也会有负marker(没有它是一种特征)。
- #寻找差异表达基因(不同的cluster的标记基因)
- #寻找cluster1的marker
- cluster1.markers <- FindMarkers(pbmc, ident.1 = 1, min.pct = 0.25)
- head(cluster1.markers, n = 5)
FindMarkers函数可以去找某一cluster与其他cluster之间相比的marker基因,ident.1表示要计算的cluster,ident.2表示所对比的其他cluster,默认为其他所有的cluster。min.pct表示基因需要在双方cluster所有细胞中表达的最小比例。
- > head(cluster1.markers, n = 5)
- p_val avg_logFC pct.1 pct.2 p_val_adj
- S100A9 0.000000e+00 3.860873 0.996 0.215 0.000000e+00
- S100A8 0.000000e+00 3.796640 0.975 0.121 0.000000e+00
- LGALS2 0.000000e+00 2.634294 0.908 0.059 0.000000e+00
- FCN1 0.000000e+00 2.352693 0.952 0.151 0.000000e+00
- CD14 2.856582e-294 1.951644 0.667 0.028 3.917516e-290
当然,也可以使用FindAllMarkers自动计算所有cluster的marker基因。
- > #cluster5对cluster0和3的marker
- > cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25)
- |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=12s
- > head(cluster5.markers, n = 5)
- p_val avg_logFC pct.1 pct.2 p_val_adj
- FCGR3A 4.742054e-206 2.949309 0.975 0.041 6.503252e-202
- CFD 1.290357e-197 2.372577 0.938 0.036 1.769595e-193
- IFITM3 1.513812e-197 2.687676 0.975 0.049 2.076042e-193
- CD68 3.196742e-194 2.096339 0.926 0.034 4.384012e-190
- RP11-290F20.3 2.790597e-189 1.886624 0.840 0.016 3.827024e-185
- > #一键自动寻找所有的marker,并且只选择positive的marker
- > pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
- Calculating cluster 0
- |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=03s
- Calculating cluster 1
- |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=09s
- Calculating cluster 2
- |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=02s
- Calculating cluster 3
- |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=03s
- Calculating cluster 4
- |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=03s
- Calculating cluster 5
- |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=13s
- Calculating cluster 6
- |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=09s
- Calculating cluster 7
- |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=10s
- Calculating cluster 8
- |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=09s
- > pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_logFC)
- # A tibble: 18 x 7
- # Groups: cluster [9]
- p_val avg_logFC pct.1 pct.2 p_val_adj cluster gene
- <dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
- 1 7.47e-120 0.770 0.917 0.587 1.02e-115 0 LDHB
- 2 2.81e- 87 0.932 0.443 0.107 3.85e- 83 0 CCR7
- 3 0. 3.86 0.996 0.215 0. 1 S100A9
- 4 0. 3.80 0.975 0.121 0. 1 S100A8
- 5 4.81e- 82 0.870 0.982 0.646 6.60e- 78 2 LTB
- 6 8.95e- 56 0.856 0.423 0.114 1.23e- 51 2 AQP3
- 7 0. 2.99 0.936 0.041 0. 3 CD79A
- 8 9.48e-271 2.49 0.622 0.022 1.30e-266 3 TCL1A
- 9 1.65e-199 2.14 0.958 0.232 2.26e-195 4 CCL5
- 10 4.93e-181 2.15 0.584 0.051 6.76e-177 4 GZMK
- 11 3.51e-184 2.30 0.975 0.134 4.82e-180 5 FCGR3A
- 12 2.03e-125 2.14 1 0.315 2.78e-121 5 LST1
- 13 1.05e-265 3.39 0.986 0.071 1.44e-261 6 GZMB
- 14 6.82e-175 3.41 0.958 0.135 9.36e-171 6 GNLY
- 15 1.48e-220 2.68 0.812 0.011 2.03e-216 7 FCER1A
- 16 1.67e- 21 1.99 1 0.513 2.28e- 17 7 HLA-DPB1
- 17 7.73e-200 5.02 1 0.01 1.06e-195 8 PF4
- 18 3.68e-110 5.94 1 0.024 5.05e-106 8 PPBP
当然,差异表达的显著性水平也可以进行检验。
- #差异表达的检验方法
- cluster1.markers <- FindMarkers(pbmc, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc", only.pos = TRUE)
这里test.use使用了ROC检验,相应的输出中包含了AUC值和power。
- > #差异表达的检验方法
- > cluster1.markers <- FindMarkers(pbmc, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc", only.pos = TRUE)
- |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=00s
- > head(cluster1.markers,5)
- myAUC avg_diff power pct.1 pct.2
- RPS6 0.831 0.4825342 0.662 1.000 0.995
- RPS12 0.830 0.5137285 0.660 1.000 0.991
- RPL32 0.823 0.4321805 0.646 0.999 0.995
- RPS27 0.819 0.4977181 0.638 0.999 0.992
- RPS14 0.817 0.4403599 0.634 1.000 0.994
接下来很自然的要去可视化这些marker,可以采用小提琴图或热图的形式来展示。
- #小提琴图可视化这些差异marker的表达
- VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
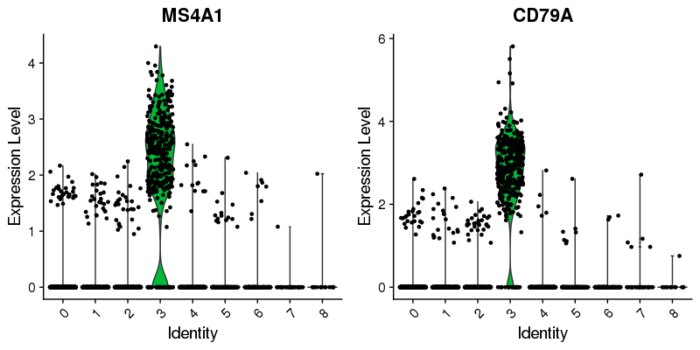
当然也可以用原始的count数(没有经过标准化和scale)来画小提琴图。
- #使用原始的count
- VlnPlot(pbmc, features = c("MS4A1", "CD79A"),slot = 'counts',log = T)
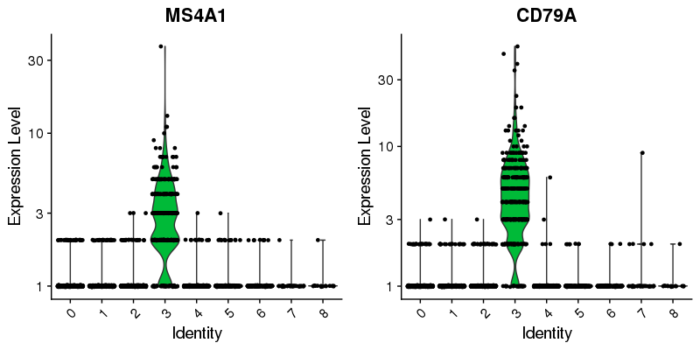
当然也能用热图的形式来展示,横坐标是细胞及其对应的cluster,纵坐标是marker基因。
- #热图的方式来表示(不展示了)
- top10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_logFC)
- DoHeatmap(pbmc, features = top10$gene) + NoLegend()
也可以具体在降维图上去看这些marker基因的表达情况。
- #在umap图上画出差异基因的表达情况
- FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A"))
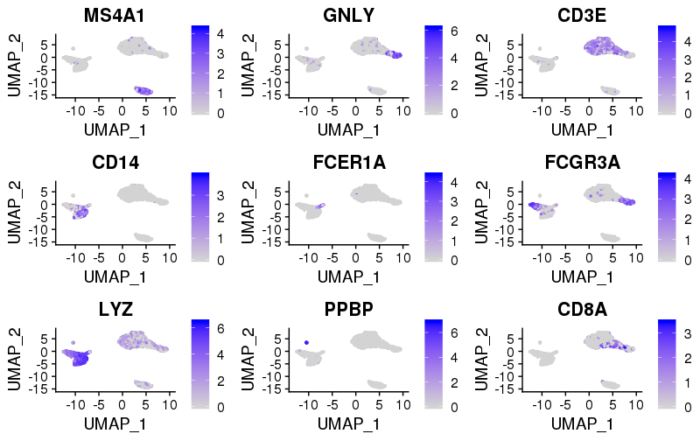
最后我们利用这些marker基因以及先验知识来identify这些cluster,并可视化。
- #为不同的cluster赋予细胞类型
- new.cluster.ids <- c("Naive CD4 T", "Memory CD4 T", "CD14+ Mono", "B", "CD8 T", "FCGR3A+ Mono", "NK", "DC", "Platelet")
- names(new.cluster.ids) <- levels(pbmc)
- pbmc <- RenameIdents(pbmc, new.cluster.ids)
- DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
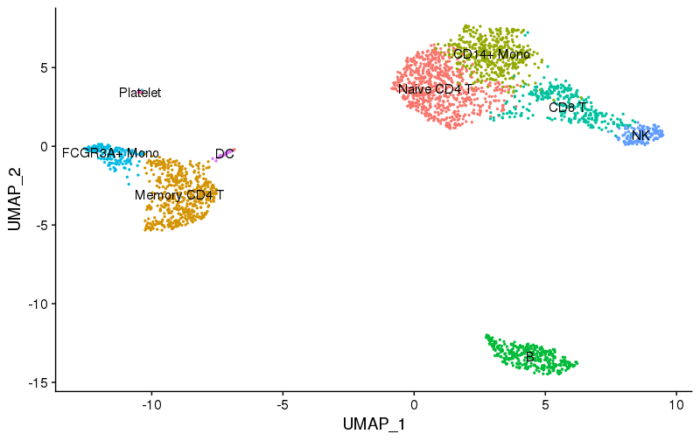
最后由于代码都是拆开来讲的,所以可能有些乱,下面贴一个完整的流程代码。
- setwd('your/path/to/seurat/filtered_gene_bc_matrices/hg19')
- #载入所需要的包
- library(dplyr)
- library(Seurat)
- #读入数据,创建seurat对象
- pbmc.data <- Read10X('./')
- pbmc <- CreateSeuratObject(counts = pbmc.data,project = 'pbmc3k',min.cells = 3,min.features = 200)
- #简单看一下seurat对象的数据结构,count matrix每一列为一个细胞,每一行为一个feature(基因)
- #这样一个count matrix存储在seurat[['RNA']]@count中,可以对应地去看一下
- pbmc.data[c("CD3D", "TCL1A", "MS4A1"), 1:30]
- pbmc[['RNA']]@counts[c("CD3D", "TCL1A", "MS4A1"),1:30]
- #两者完全一致
- #质控,包括对每个细胞检测到的基因数,以及线粒体基因等进行质控
- #线粒体基因质控
- pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
- VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
- #质控reads数和基因数的关系
- plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
- plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
- CombinePlots(plots = list(plot1, plot2))
- #挑选基因数大于200小于2500,线粒体基因数小于5%的数据(data-selection)
- pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
- #再来看下小提琴图
- VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
- #筛选完数据,进行标准化
- pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
- #这个scale factor就是默认值可以不改
- #寻找差异表达基因(细胞与细胞之间方差大)这也很好理解,基因的个数可以根据数据和后续分析进行调整
- pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
- #来看下这些差异表达的基因
- #数据存储在seurat[['RNA']]@var.features,可以用VariableFeatures函数进行调用
- top10 <- head(VariableFeatures(pbmc), 10)
- plot1 <- VariableFeaturePlot(pbmc)
- plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
- CombinePlots(plots = list(plot1, plot2))
- #scale,feature均值中心化为0,方差为1的归一化操作,降低一些高表达feature的主导程度
- #数据存储在seurat[['RNA']]@scale.data
- all.genes <- rownames(pbmc)
- pbmc <- ScaleData(pbmc, features = all.genes)
- #PCA降维,高维向量是所选择的2000个高差异表达基因
- pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
- print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
- #可视化
- VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
- DimPlot(pbmc, reduction = "pca")
- #用heatmap查看每一个PC上异质性的来源,横轴是选择的最‘极端’的细胞
- DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)
- DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)
- #单细胞RNA-seq的噪声大,因此采用PCA值作为聚类的依据,那么如何判断采用哪些PC或多少PC?
- #JackStraw法
- pbmc <- JackStraw(pbmc, num.replicate = 100)
- pbmc <- ScoreJackStraw(pbmc, dims = 1:20)
- JackStrawPlot(pbmc, dims = 1:15)
- #elbowplot
- ElbowPlot(pbmc)
- #最终选择前10个pc作为聚类的依据
- #resolution的参数可以具体调节
- pbmc <- FindNeighbors(pbmc, dims = 1:10)
- pbmc <- FindClusters(pbmc, resolution = 0.5)
- #查看前5个细胞的聚类情况
- head(Idents(pbmc), 5)
- #非线性降维tSNE或UMAP
- pbmc <- RunUMAP(pbmc, dims = 1:10)
- DimPlot(pbmc, reduction = "umap")
- pbmc <- RunTSNE(pbmc, dims = 1:10)
- DimPlot(pbmc, reduction = "tsne")
- #寻找差异表达基因(不同的cluster的标记基因)
- #寻找cluster1的marker
- cluster1.markers <- FindMarkers(pbmc, ident.1 = 1, min.pct = 0.25)
- head(cluster1.markers, n = 5)
- #cluster5对cluster0和3的marker
- cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25)
- head(cluster5.markers, n = 5)
- #一键自动寻找所有的marker,并且只选择positive的marker
- pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
- pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_logFC)
- #差异表达的检验方法
- cluster1.markers <- FindMarkers(pbmc, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc", only.pos = TRUE)
- head(cluster1.markers,5)
- #可视化这些差异marker的表达
- VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
- #使用原始的count
- VlnPlot(pbmc, features = c("MS4A1", "CD79A"),slot = 'counts',log = T)
- #在umap图上画出差异基因的表达情况
- FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A"))
- #热图的方式来表示(不展示了)
- top10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_logFC)
- DoHeatmap(pbmc, features = top10$gene) + NoLegend()
- #为不同的cluster赋予细胞类型
- new.cluster.ids <- c("Naive CD4 T", "Memory CD4 T", "CD14+ Mono", "B", "CD8 T", "FCGR3A+ Mono", "NK", "DC", "Platelet")
- names(new.cluster.ids) <- levels(pbmc)
- pbmc <- RenameIdents(pbmc, new.cluster.ids)
- DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
OK,今天利用seurat进行单细胞RNA-seq聚类的分析流程就分享到这里,后面还会利用seurat做更多的单细胞数据分析。