

单细胞RNA-seq很大的特点是数据量大,数据噪声高。因此在对一些特别小的类群进行分析,或者增加数据量降噪的情况下,我们往往需要将多个单细胞RNA-seq的数据集整合起来,这些数据集可能来自不同的研究者,可能使用了不同的测序技术等等,因此将这些数据整合起来的困难点就在于如何去除不同数据集之间的批次效应。Seurat提供了自带的多种整合单细胞数据集的方法,这里记录一下使用流程。Seurat进行数据整合的思路也很简单,先去寻找不同数据集之间的锚点(anchor),这些anchor反映了不同数据集中‘我们假定是同样的生物学状态的’各个细胞之间的相关性,再利用这种相关性对表达矩阵进行修正从而将多个数据集整合起来,或者作为预测器发挥作用。Seurat提供了多种整合分析的方法,下面一一结合代码演示。
- 标准流程
标准流程主要可以实现两个功能,一是将多组单细胞RNA-seq数据整合起来,二是作为预测器,将一个已经定义好的参考数据库中的细胞标签,转移到一个新的数据集中,相当于用参考数据库对新数据中的cell类型进行分类。其思路都是一致的,都是先寻找anchor,再根据anchor进行整合或分类,这里值得一提的是,在整合过程中seurat会对表达矩阵进行修正,而在预测过程中则不会对表达矩阵进行处理,仅仅是预测各个细胞的细胞类型。标准流程使用通过四种不同测序技术得到的人类胰岛单细胞数据集,其中”celseq”, “celseq2”, “smartseq2″三组数据用于整合成一个大的reference,”fluidigmc1″用于预测细胞类型,这些数据都已经整合到SeuratData包中。
- setwd('/your/path/to/seurat')
- #加载所需要的包
- library(Seurat)
- library(SeuratData)
- #数据已经整合到SeuratData包中
- #InstallData("panc8")
- #将数据按技术进行拆分
- data("panc8")
- pancreas.list <- SplitObject(panc8, split.by = "tech")
- pancreas.list <- pancreas.list[c("celseq", "celseq2", "fluidigmc1", "smartseq2")]
通过默认函数的拆分,pancrease.list中包含了4个seurat对象。很自然的,第一步进行标准化,并寻找各个数据集的差异表达基因各2000个。
- #在寻找anchor之前先进行标准化,找出各个数据集自身的差异表达基因
- for (i in 1:length(pancreas.list)) {
- pancreas.list[[i]] <- NormalizeData(pancreas.list[[i]], verbose = FALSE)
- pancreas.list[[i]] <- FindVariableFeatures(pancreas.list[[i]], selection.method = "vst",
- nfeatures = 2000, verbose = FALSE)
- }
做完标准化后,即可用FindIntegrationAnchors函数去寻找用于整合的anchor。
- #寻找anchor,这里只用了其中的三个数据,另一个数据会在后面用到
- reference.list <- pancreas.list[c("celseq", "celseq2", "smartseq2")]
- pancreas.anchors <- FindIntegrationAnchors(object.list = reference.list, anchor.features = 2000,dims = 1:30)
说下参数的意思,anchor.feature表示在寻找anchor过程中所设定的feature数,这个值也直接决定了整合后的表达矩阵中有多少个基因。dims表示在CCA的过程中选择哪些维度进行后续的计算,这里选择了默认参数,当然也可以进行调整,官网推荐值在10-50之间。用这些anchor去矫正技术和批次差异。
- #利用这样的anchor去矫正批次和技术差异(相当于找到了共同的锚点,用这样的锚点去矫正表达水平)
- pancreas.integrated <- IntegrateData(anchorset = pancreas.anchors, dims = 1:30)
看一下数据结构,IntegrateData这个函数可以将锚点信息转换成整合的表达矩阵并以seurat对象的方式存储,在assay中可以看到,除了整合之前的表达矩阵存储在RNA中,还多了整合后的表达矩阵存储在integrated中,整合后的表达矩阵中只有相应数量的基因(feature)。这两个表达矩阵可以通过DefaultAssay函数进行切换。
- #将seurat对象的默认assay设置成整合后的表达矩阵
- DefaultAssay(pancreas.integrated) <- "integrated"
这个新的整合后的表达矩阵就可以进行一系列的下游分析了,参考seurat进行单细胞RNA-seq聚类分析。那么老规矩,将这个整合好的数据集按细胞类型(非技术类型)进行聚类,再用tSNE或UMAP进行可视化。OK,那么我们就按照之前的教程再重新跑一遍,相当于是回顾,先scale数据,需要注意一点,ScaleData函数在scale过程中默认取差异表达基因进行scale,但由于这里整合好的表达矩阵只有相应的差异表达基因,因此不用单独设置取全基因进行scale。
- #载入包
- library(ggplot2)
- library(cowplot)
- #归一化,注意,这里只有2000个基因
- pancreas.integrated <- ScaleData(pancreas.integrated, verbose = FALSE)
归一化后进行PCA降维,注意RunPCA函数默认取50个PC轴,这里取了30个,并将这全部30个用于下游的tSNE或者UMAP。
- #PCA
- pancreas.integrated <- RunPCA(pancreas.integrated, npcs = 30, verbose = FALSE)
进行UMAP降维可视化,选择了PCA中的全部30个PC作为高维向量。
- #使用umap根据pca降维的情况,取其前30个(总共也就30个)PC作为降维的高维向量
- pancreas.integrated <- RunUMAP(pancreas.integrated, reduction = "pca", dims = 1:30)
- #可视化,分别以技术和细胞类型作为标签
- p1 <- DimPlot(pancreas.integrated, reduction = "umap", group.by = "tech")
- p2 <- DimPlot(pancreas.integrated, reduction = "umap", group.by = "celltype", label = TRUE,
- repel = TRUE) NoLegend()
- plot_grid(p1, p2)
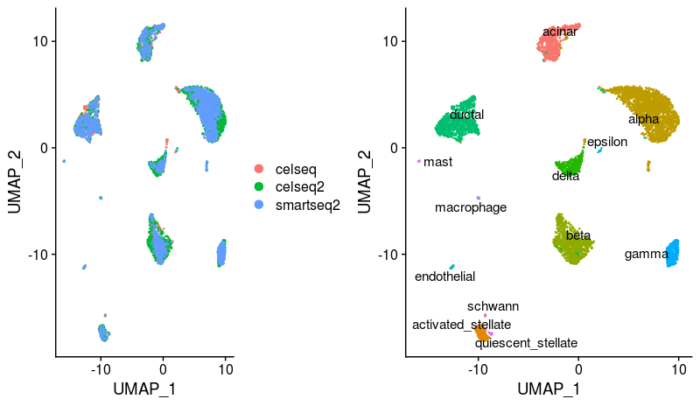
稍微提一句,这里的聚类不是通过聚类算法算出来的,而是在metadata中每个细胞就已经标上了细胞类型,tech和celltype的信息都存储在metadata中。类似的,也可以基于anchor实现分类器(label transfer)。需要注意的是,与整合不同,分类器只是预测每个细胞在参考数据中的细胞类型,并不会对表达矩阵进行修正。这里参考数据集选择上面整合好的数据,query数据集选择之前提到的未使用的数据。
- #类似的,也可以基于anchor实现分类器(label transfer)
- #需要注意的是,与整合不同,分类器只是预测每个细胞在参考数据中的细胞类型,并不会对表达矩阵进行修正
- #这里使用之前还未使用的另一组数据
- pancreas.query <- pancreas.list[["fluidigmc1"]]
- pancreas.anchors <- FindTransferAnchors(reference = pancreas.integrated, query = pancreas.query,
- dims = 1:30)
- #这里寻找transfer anchor有两种方法,一种是基于PCA的方法,一种是基于CCA的方法,默认且官方推荐PCA的方法
注意,FindTransferAnchors函数与寻找整合的anchor函数不太一样的地方在于他一般不需要设置feature。TransferData函数作为分类机,可以基于anchor的模型进行分类,得到预测结果。
- #TransferData函数作为分类机,可以基于anchor的模型进行分类,得到预测结果
- predictions <- TransferData(anchorset = pancreas.anchors, refdata = pancreas.integrated$celltype,
- dims = 1:30)
- #将预测结果加入metadata
- pancreas.query <- AddMetaData(pancreas.query, metadata = predictions)
查看分类的准确性。
- #查看预测准确率
- prediction.match <- pancreas.query$predicted.id == pancreas.query$celltype
- table(prediction.match)
结果如下:
- > table(prediction.match)
- prediction.match
- FALSE TRUE
- 18 620
准确率97.18%,很漂亮的数字了。另外,根据官方的说法,即使测试集中只有少数几个细胞代表某种细胞类型,可以用这种方法进行预测,比如epsilon。
- > table(pancreas.query$predicted.id)
- acinar activated_stellate alpha beta
- 21 17 248 258
- delta ductal endothelial epsilon
- 22 33 13 1
- gamma macrophage mast schwann
- 17 1 2 5
最后,我们进一步通过marker基因的表达来判断预测的准确性。
- #进一步通过marker基因的表达来判断预测的准确性
- VlnPlot(pancreas.query, c("REG1A", "PPY", "SST", "GHRL", "VWF", "SOX10"), group.by = "predicted.id")
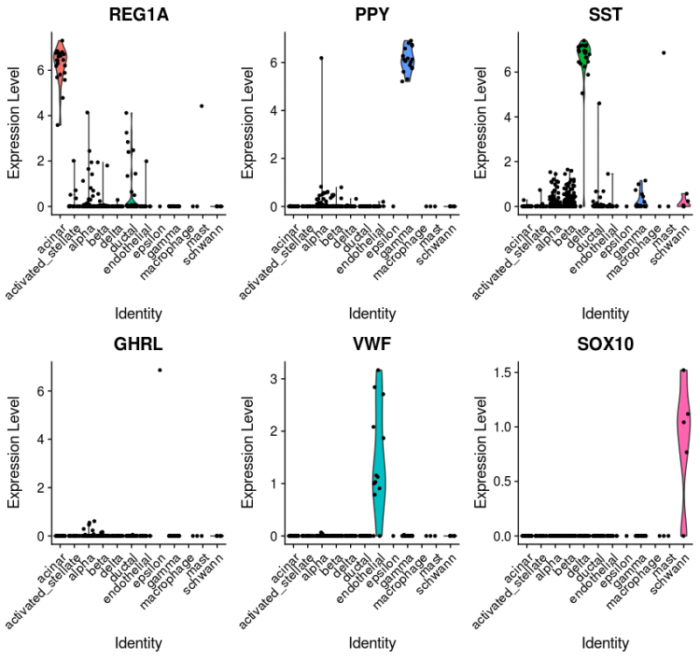
最后老规矩,贴一个完整代码。
- setwd('/your/path/to/seurat')
- #加载所需要的包
- library(Seurat)
- library(SeuratData)
- #先尝试标准工作流程,后面尝试其他的高级姿势
- #标准工作流程,可以实现两个目标
- #1.将多个不同的单细胞RNA-seq数据整合成一个大的reference
- #2.作为分类器,将一个已经定义好的参考数据库中的细胞标签,转移到一个新的数据集中,相当于用参考数据库对新数据中的cell类型进行分类
- #数据已经整合到SeuratData包中
- #标准流程是将使用四种不同测序技术得到的人类胰岛细胞数据整合起来
- #InstallData("panc8")
- #将数据按技术进行拆分
- data("panc8")
- pancreas.list <- SplitObject(panc8, split.by = "tech")
- pancreas.list <- pancreas.list[c("celseq", "celseq2", "fluidigmc1", "smartseq2")]
- #来看下数据结构,pancrease.list包含了4个seurat对象
- #流程
- #在寻找anchor之前先进行标准化,找出各个数据集自身的差异表达基因
- for (i in 1:length(pancreas.list)) {
- pancreas.list[[i]] <- NormalizeData(pancreas.list[[i]], verbose = FALSE)
- pancreas.list[[i]] <- FindVariableFeatures(pancreas.list[[i]], selection.method = "vst",
- nfeatures = 2000, verbose = FALSE)
- }
- #寻找anchor,这里只用了其中的三个数据,另一个数据会在后面用到
- reference.list <- pancreas.list[c("celseq", "celseq2", "smartseq2")]
- pancreas.anchors <- FindIntegrationAnchors(object.list = reference.list, anchor.features = 2000, dims = 1:30)
- #dims取了默认参数,也可以进行调整,官网推荐值在10-50之间
- #利用这样的anchor去矫正批次和技术差异(相当于找到了共同的锚点,用这样的锚点去矫正表达水平)
- pancreas.integrated <- IntegrateData(anchorset = pancreas.anchors, dims = 1:30)
- #看一下数据结构,IntegrateData这个函数可以将锚点信息转换成整合的表达矩阵,在assay中可以看到,除了整合之前的表达矩阵存储在RNA中,还多了整合后的表达矩阵存储在integrated中
- #这两个表达矩阵可以通过DefaultAssay函数进行切换
- #另外值得注意的是,在整合后的表达矩阵中,只有2000行,对应了所找到的差异表达基因
- #这个新的整合后的表达矩阵就可以进行一系列的下游分析了
- #老规矩,按细胞类型(非技术类型)进行聚类,再用tSNE或者UMAP可视化
- library(ggplot2)
- library(cowplot)
- #将seurat对象的默认assay设置成整合后的表达矩阵
- DefaultAssay(pancreas.integrated) <- "integrated"
- #归一化,注意,这里只有2000个基因
- pancreas.integrated <- ScaleData(pancreas.integrated, verbose = FALSE)
- #PCA
- pancreas.integrated <- RunPCA(pancreas.integrated, npcs = 30, verbose = FALSE)
- #使用umap根据pca降维的情况,取其前30个(总共也就30个)PC作为降维的高维向量
- pancreas.integrated <- RunUMAP(pancreas.integrated, reduction = "pca", dims = 1:30)
- #可视化,分别以技术和细胞类型作为标签
- p1 <- DimPlot(pancreas.integrated, reduction = "umap", group.by = "tech")
- p2 <- DimPlot(pancreas.integrated, reduction = "umap", group.by = "celltype", label = TRUE,
- repel = TRUE) NoLegend()
- plot_grid(p1, p2)
- #tech和celltype都储存在metadata中
- #类似的,也可以基于anchor实现分类器(label transfer)
- #需要注意的是,与整合不同,分类器只是预测每个细胞在参考数据中的细胞类型,并不会对表达矩阵进行修正
- #这里使用之前还未使用的另一组数据
- pancreas.query <- pancreas.list[["fluidigmc1"]]
- pancreas.anchors <- FindTransferAnchors(reference = pancreas.integrated, query = pancreas.query,
- dims = 1:30)
- #这里寻找transfer anchor有两种方法,一种是基于PCA的方法,一种是基于CCA的方法,默认且官方推荐PCA的方法
- #TransferData函数作为分类机,可以基于anchor的模型进行分类,得到预测结果
- predictions <- TransferData(anchorset = pancreas.anchors, refdata = pancreas.integrated$celltype,
- dims = 1:30)
- #将预测结果加入metadata
- pancreas.query <- AddMetaData(pancreas.query, metadata = predictions)
- #查看预测准确率
- prediction.match <- pancreas.query$predicted.id == pancreas.query$celltype
- table(prediction.match)
- #准确率97.18%
- table(pancreas.query$predicted.id)
- #根据官方的说法,即使测试集中只有少数几个细胞代表某种细胞类型,可以用这种方法进行预测,比如epsilon
- #进一步通过marker基因的表达来判断预测的准确性
- VlnPlot(pancreas.query, c("REG1A", "PPY", "SST", "GHRL", "VWF", "SOX10"), group.by = "predicted.id")
- SC Transform
SC Transform相当于是标准流程的一种modified版本,其原理和标准流程类似,只不过标准流程中使用log函数对表达矩阵进行标准化,这里使用SC transform对表达矩阵进行标准化,再对皮尔逊残差进行协调整合。直接上代码:
- setwd('/your/path/to/seurat')
- #原理和标准流程是类似的,只不过标准流程中使用log函数对表达矩阵进行标准化,这里使用SC transform对表达矩阵进行标准化,再对皮尔逊残差进行协调整合
- #加载包
- library(Seurat)
- library(ggplot2)
- library(SeuratData)
- #可能是数据比较大吧,他这里设置了一下全局变量的最大容量
- options(future.globals.maxSize = 4000 * 1024^2)
- #还是之前的4组数据
- data("panc8")
- pancreas.list <- SplitObject(panc8, split.by = "tech")
- pancreas.list <- pancreas.list[c("celseq", "celseq2", "fluidigmc1", "smartseq2")]
这里还是用的标准流程中的4组数据,官网还提供了8组数据的一个练手,但我发现这种方法的计算开销很大,跑的时间也很长,所以我就没有去实战,建议直接用R去跑前期的数据处理,存个RData,后期的可视化可以用Rstudio,这样会节省一些时间。那么第一步肯定还是标准化,在标准流程中用了NormalizeData函数,默认取logNormalize,这里使用SC transform进行标准化。
- #第一步肯定还是标准化,在标准流程中用了NormalizeData函数,默认取logNormalize,这里使用ST transform进行标准化
- for (i in 1:length(pancreas.list)) {
- pancreas.list[[i]] <- SCTransform(pancreas.list[[i]], verbose = T)
- }
- #因为用时较长,建议把verbose设置成True
选择用于下游寻找anchor并整合的feature,这里选择3000个基因。
- #选择用于下游寻找anchor并整合的feature,这里选择3000个基因
- pancreas.features <- SelectIntegrationFeatures(object.list = pancreas.list, nfeatures = 3000)
整合前预处理,保证所有必要的皮尔逊残差都被计算到。
- #整合前预处理,保证所有必要的皮尔逊残差都被计算到
- pancreas.list <- PrepSCTIntegration(object.list = pancreas.list, anchor.features = pancreas.features, verbose = T)
整合步骤与标准流程类似,不过需要注意标准化方法选择SCT。
- #整合步骤与标准流程类似,不过需要注意标准化方法选择SCT
- pancreas.anchors <- FindIntegrationAnchors(object.list = pancreas.list, normalization.method = "SCT",
- anchor.features = pancreas.features, verbose = T)
- pancreas.integrated <- IntegrateData(anchorset = pancreas.anchors, normalization.method = "SCT",
- verbose = T)
那么和标准流程一样的,下面就可以进行各种下游分析了,不过有一个注意点是SCT整合后就不要用ScaleData进行scale了。
- #那么和标准流程一样的,下面就可以进行各种下游分析了
- #有一个注意点是SCT整合后就不要用ScaleData进行scale了
- DefaultAssay(pancreas.integrated) <- "integrated"
- pancreas.integrated <- RunPCA(pancreas.integrated, verbose = FALSE)
- pancreas.integrated <- RunUMAP(pancreas.integrated, dims = 1:30)
- plots <- DimPlot(pancreas.integrated, group.by = c("tech", "celltype"), combine = FALSE)
- plots <- lapply(X = plots, FUN = function(x) x theme(legend.position = "top") guides(color = guide_legend(nrow = 3, byrow = TRUE, override.aes = list(size = 3))))
- CombinePlots(plots)
- #这画图操作可真骚
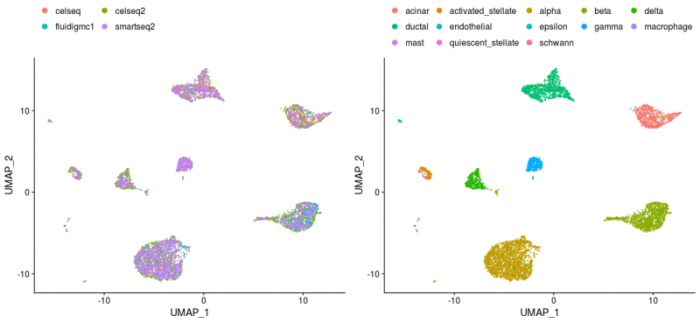
可以看到,大部分细胞也都按照各自的label与其他细胞类群区分了开来,说明合并是有效的。最后贴个完整的代码:
- setwd('/your/path/to/seurat')
- #原理和标准流程是类似的,只不过标准流程中使用log函数对表达矩阵进行标准化,这里使用ST transform对表达矩阵进行标准化,再对皮尔逊残差进行协调整合
- #加载包
- library(Seurat)
- library(ggplot2)
- library(SeuratData)
- #可能是数据比较大吧,他这里设置了一下全局变量的最大容量
- options(future.globals.maxSize = 4000 * 1024^2)
- #还是之前的4组数据
- data("panc8")
- pancreas.list <- SplitObject(panc8, split.by = "tech")
- pancreas.list <- pancreas.list[c("celseq", "celseq2", "fluidigmc1", "smartseq2")]
- #第一步肯定还是标准化,在标准流程中用了NormalizeData函数,默认取logNormalize,这里使用ST transform进行标准化
- for (i in 1:length(pancreas.list)) {
- pancreas.list[[i]] <- SCTransform(pancreas.list[[i]], verbose = T)
- }
- #因为用时较长,建议把verbose设置成True
- #选择用于下游寻找anchor并整合的feature,这里选择3000个基因
- pancreas.features <- SelectIntegrationFeatures(object.list = pancreas.list, nfeatures = 3000)
- #整合前预处理,保证所有必要的皮尔逊残差都被计算到
- pancreas.list <- PrepSCTIntegration(object.list = pancreas.list, anchor.features = pancreas.features, verbose = T)
- #整合步骤与标准流程类似,不过需要注意标准化方法选择SCT
- pancreas.anchors <- FindIntegrationAnchors(object.list = pancreas.list, normalization.method = "SCT",
- anchor.features = pancreas.features, verbose = T)
- pancreas.integrated <- IntegrateData(anchorset = pancreas.anchors, normalization.method = "SCT",
- verbose = T)
- #那么和标准流程一样的,下面就可以进行各种下游分析了
- #有一个注意点是SCT整合后就不要用ScaleData进行scale了
- DefaultAssay(pancreas.integrated) <- "integrated"
- pancreas.integrated <- RunPCA(pancreas.integrated, verbose = FALSE)
- pancreas.integrated <- RunUMAP(pancreas.integrated, dims = 1:30)
- plots <- DimPlot(pancreas.integrated, group.by = c("tech", "celltype"), combine = FALSE)
- plots <- lapply(X = plots, FUN = function(x) x theme(legend.position = "top") guides(color = guide_legend(nrow = 3, byrow = TRUE, override.aes = list(size = 3))))
- CombinePlots(plots)
- #这画图操作可真骚
- #可以看到,大部分细胞也都按照各自的label与其他细胞类群区分了开来,说明合并是有效的
- Reference-based
Reference-based integration也是标准流程的一种modified版本,在之前的分析中,无论是标准流程还是SC Transform,在寻找anchor的过程中都要在数据集之间进行两两比较,每个数据集在下游分析中都有相同的权重,因此其计算开销是非常大的,在上面的SC Transform中我也有所感触。那么reference-based integration的想法就是,把其中的一个或几个数据集设置成reference,那么在寻找anchor的时候就不需要将所有的数据集两两比较了,而只需要将query数据集和reference数据集一一比较即可,从而大大减小了计算量。比如我有10个数据集,那么两两比较需要计算45次,但如果把一个数据集设置成reference,则只需要将剩下的9个数据集和reference数据集比较9次即可。因此,这种reference-based integration适用于数据集较多的情况下的整合,并且对于log标准化还是SC Transform标准化也都是适用的。
事实上这种reference-based integration也不需要使用者事先选择参考数据集,一般来说尽量选择细胞数目较多,敏感性较好的数据集作为参考数据集即可,选择不同的参考数据集可能会对最终的结果有轻微的影响。下面的演示选择的数据是使用9种技术制备的人类PBMC数据集,可以在SeuratData包中找到,其中10X v3 dataset作为参考数据集。
- setwd('/your/path/to/seurat')
- #reference-based integration和标准流程也是类似的,其区别在于可以将其中的一个或几个数据集设置成reference,从而减少计算量
- #数据使用的是用8种技术制备的人类PBMC数据集,可以在SeuratData包中找到
- #载入包
- library(Seurat)
- library(SeuratData)
- library(ggplot2)
- #载入人类PBMC数据集
- data("pbmcsca")
- pbmc.list <- SplitObject(pbmcsca, split.by = "Method")
加载完数据后进行标准化,这里选择SC Transform进行标准化,由于有9组数据,所以跑的时间也会比较久。
- #标准化,使用SC Transform
- for (i in names(pbmc.list)) {
- pbmc.list[[i]] <- SCTransform(pbmc.list[[i]], verbose = T)
- }
选择用于寻找anchor和下游分析的feature,这里仍然选择3000个基因,挑选完基因后,即可对相应的皮尔逊残差进行计算,完成整合前的预处理。
- #选择用于寻找anchor和下游分析的feature,这里仍然选择3000个基因
- #挑选完基因后,即可对相应的皮尔逊残差进行计算,完成整合前的预处理
- pbmc.features <- SelectIntegrationFeatures(object.list = pbmc.list, nfeatures = 3000)
- pbmc.list <- PrepSCTIntegration(object.list = pbmc.list, anchor.features = pbmc.features)
选择参考数据集,可以指定一个或多个数据集作为参考数据集。
- #选择参考数据集
- reference_dataset <- which(names(pbmc.list) == "10x Chromium (v3)")
- #这里的返回值是5,如果想设置几个数据集为一个reference,可以使用c(5,6)等类似的写法
计算anchor并整合,与SC Transform唯一的区别在于多了reference参数。
- #计算anchor并整合,与SC Transform唯一的区别在于多了reference参数
- pbmc.anchors <- FindIntegrationAnchors(object.list = pbmc.list, normalization.method = "SCT", anchor.features = pbmc.features, reference = reference_dataset)
- pbmc.integrated <- IntegrateData(anchorset = pbmc.anchors, normalization.method = "SCT")
感觉anchor的计算速度显著提升了,但整合的计算开销依然很大。接着还是像上面的流程一样,画出UMAP图实现可视化。
- #可视化
- DefaultAssay(pancreas.integrated) <- "integrated"
- pbmc.integrated <- RunPCA(object = pbmc.integrated, verbose = T)
- pbmc.integrated <- RunUMAP(object = pbmc.integrated, dims = 1:30)
- plots <- DimPlot(pbmc.integrated, group.by = c("Method", "CellType"), combine = FALSE)
- plots <- lapply(X = plots, FUN = function(x) x theme(legend.position = "top") guides(color = guide_legend(nrow = 4, byrow = TRUE, override.aes = list(size = 2.5))))
- CombinePlots(plots)
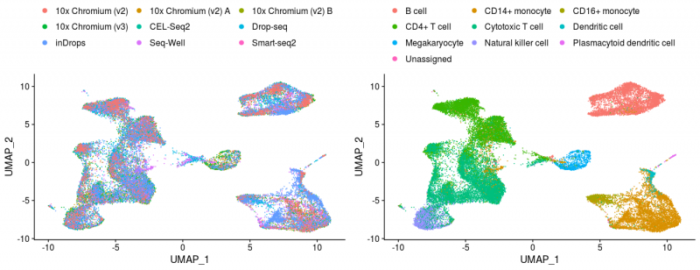
按照技术将图片分开,可以看到,使用不同的技术,其聚类的pattern是一致的。
- #按照技术将图片拆分
- DimPlot(pbmc.integrated, group.by = "CellType", split.by = "Method", ncol = 3)
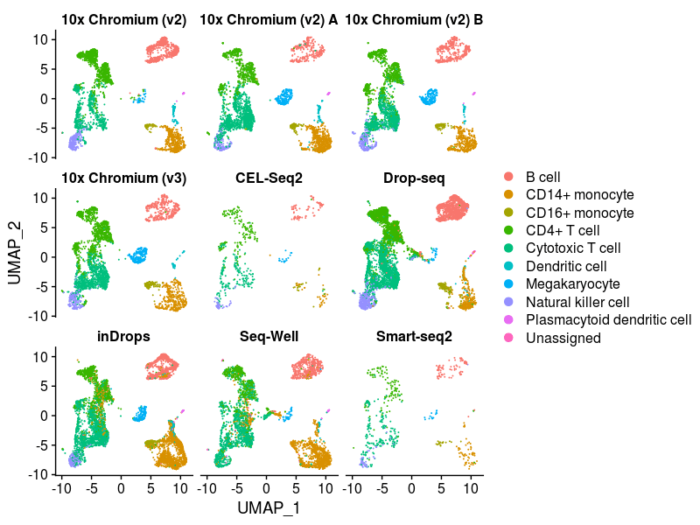
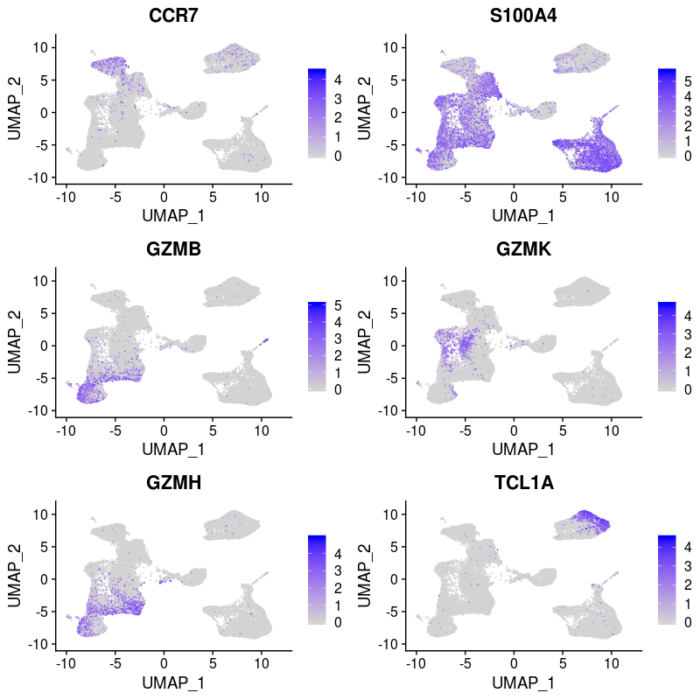
最后使用maker基因进一步验证聚类的情况。
- #使用maker基因的表达量进一步验证聚类情况
- DefaultAssay(pbmc.integrated) <- "RNA"
- #对RNA中存储的原始表达矩阵进行标准化
- pbmc.integrated <- NormalizeData(pbmc.integrated, verbose = T)
- FeaturePlot(pbmc.integrated, c("CCR7", "S100A4", "GZMB", "GZMK", "GZMH", "TCL1A"))
- #FeaturePlot也可以用method进行split
完整代码如下:
- setwd('/your/path/to/seurat')
- #reference-based integration和标准流程也是类似的,其区别在于可以将其中的一个或几个数据集设置成reference,从而减少计算量
- #数据使用的是用8种技术制备的人类PBMC数据集,可以在SeuratData包中找到
- #载入包
- library(Seurat)
- library(SeuratData)
- library(ggplot2)
- #载入人类PBMC数据集
- data("pbmcsca")
- pbmc.list <- SplitObject(pbmcsca, split.by = "Method")
- #标准化,使用SC Transform
- for (i in names(pbmc.list)) {
- pbmc.list[[i]] <- SCTransform(pbmc.list[[i]], verbose = T)
- }
- #选择用于寻找anchor和下游分析的feature,这里仍然选择3000个基因
- #挑选完基因后,即可对相应的皮尔逊残差进行计算,完成整合前的预处理
- pbmc.features <- SelectIntegrationFeatures(object.list = pbmc.list, nfeatures = 3000)
- pbmc.list <- PrepSCTIntegration(object.list = pbmc.list, anchor.features = pbmc.features)
- #选择参考数据集
- reference_dataset <- which(names(pbmc.list) == "10x Chromium (v3)")
- #这里的返回值是5,如果想设置几个数据集为一个reference,可以使用c(5,6)等类似的写法
- #计算anchor并整合,与SC Transform唯一的区别在于多了reference参数
- pbmc.anchors <- FindIntegrationAnchors(object.list = pbmc.list, normalization.method = "SCT",
- anchor.features = pbmc.features, reference = reference_dataset)
- pbmc.integrated <- IntegrateData(anchorset = pbmc.anchors, normalization.method = "SCT")
- #可视化
- DefaultAssay(pancreas.integrated) <- "integrated"
- pbmc.integrated <- RunPCA(object = pbmc.integrated, verbose = T)
- pbmc.integrated <- RunUMAP(object = pbmc.integrated, dims = 1:30)
- plots <- DimPlot(pbmc.integrated, group.by = c("Method", "CellType"), combine = FALSE)
- plots <- lapply(X = plots, FUN = function(x) x theme(legend.position = "top") guides(color = guide_legend(nrow = 4,
- byrow = TRUE, override.aes = list(size = 2.5))))
- CombinePlots(plots)
- #按照技术将图片拆分
- DimPlot(pbmc.integrated, group.by = "CellType", split.by = "Method", ncol = 3)
- #使用maker基因的表达量进一步验证聚类情况
- DefaultAssay(pbmc.integrated) <- "RNA"
- #对RNA中存储的原始表达矩阵进行标准化
- pbmc.integrated <- NormalizeData(pbmc.integrated, verbose = T)
- FeaturePlot(pbmc.integrated, c("CCR7", "S100A4", "GZMB", "GZMK", "GZMH", "TCL1A"))
- #FeaturePlot也可以用method进行split
- Reciprocal PCA
在之前的流程中,FindIntegrationAnchors函数中,我们降维方法都默认选择了CCA,但对于较大的数据集来说,CCA对计算资源的开销特别大,会成为‘限速’的卡点。因此这里就介绍了一种替代方案:通过reciprocal PCA(反式PCA)来确定寻找anchor的有效空间。其基本原理是,对每个数据集进行PCA主成分分析,再将两两数据集的PCA空间投影到一起,那么这两个数据集之间的anchor就被约束在了满足相同互领域的空间中,从而大大减少了搜索范围和计算量。当然,使用rPCA的方法进行整合,也可以引入reference来进一步降低计算量。
一般来说,reciprocal PCA得出的整合结果与CCA得出的整合结果基本相似,但计算量得以大大减少。但如果数据集之间的异质性特别高(比如物种之间的整合),由于只有一小部分feature能用于整合,因此CCA的结果可能会更好。下面的演示使用SeuratData包提供的免疫细胞图谱数据,共8个供体,每个供体down sample 5000个细胞,共40k个细胞。
- setwd('/your/path/to/seurat')
- #使用reciprocal PCA的方法寻找数据集之间的anchor,从而进行整合
- #数据使用SeuratData提供的人类免疫细胞图谱的数据集的downsample,共40k个细胞
- #没有用完整数据是因为我根据提示找不到原始数据,我好菜
- #加载包
- library(Seurat)
- library(SeuratData)
- library(ggplot2)
- #用于并行计算的包
- library(future)
- library(future.apply)
- #初始化设置
- plan("multiprocess", workers = 4)
- options(future.globals.maxSize = 20000 * 1024^2)
- #加载数据
- # AvailableData()
- # InstallData("hcabm40k.SeuratData")
- data(hcabm40k)
这个并行计算我觉得很香,后面会考虑仔细研究一下专门做一期R语言的并行计算。加载完数据后进行标准化,并寻找各个数据集的差异表达基因,我猜测这里寻找差异表达基因是为了减少后面scale的计算量。
- #将数据按来源拆分,并行计算标准化并寻找差异表达基因各2000个
- bm40k.list <- SplitObject(hcabm40k, split.by = "orig.ident")
- bm40k.list <- lapply(X = bm40k.list, FUN = function(x) {
- x <- NormalizeData(x, verbose = FALSE)
- x <- FindVariableFeatures(x, verbose = FALSE)
- })
- #我猜测在这里运行FindVariableFeatures是为了节省下面scale步骤中的时间,因为scale默认只scale差异表达基因
挑选用于寻找anchor并整合的feature。
- #挑选用于整合的基因
- features <- SelectIntegrationFeatures(object.list = bm40k.list)
Reciprocal PCA整合前,先对每个数据集进行PCA主成分分析,注意在跑PCA之前要先scale。
- #rPCA整合前,先scale再做PCA
- bm40k.list <- lapply(X = bm40k.list, FUN = function(x) {
- x <- ScaleData(x, features = features, verbose = FALSE)
- x <- RunPCA(x, features = features, verbose = FALSE)
- })
使用一个男人和一个女人的数据作为reference进行整合,注意降维选择rpca。这里选择了前1-30个维度,随着数据量的增大,异质性的提高,可以适当增加用于整合的维度。
- #使用一个男人和一个女人的数据作为reference进行整合,注意降维选择rpca
- anchors <- FindIntegrationAnchors(object.list = bm40k.list, reference = c(1, 2), reduction = "rpca", dims = 1:30)
- bm40k.integrated <- IntegrateData(anchorset = anchors, dims = 1:30)
整合完后就可以进行下游可视化分析了,这里不再赘述。
- #下游分析与可视化
- bm40k.integrated <- ScaleData(bm40k.integrated, verbose = T)
- bm40k.integrated <- RunPCA(bm40k.integrated, verbose = T)
- bm40k.integrated <- RunUMAP(bm40k.integrated, dims = 1:30)
- DimPlot(bm40k.integrated, group.by = "orig.ident")
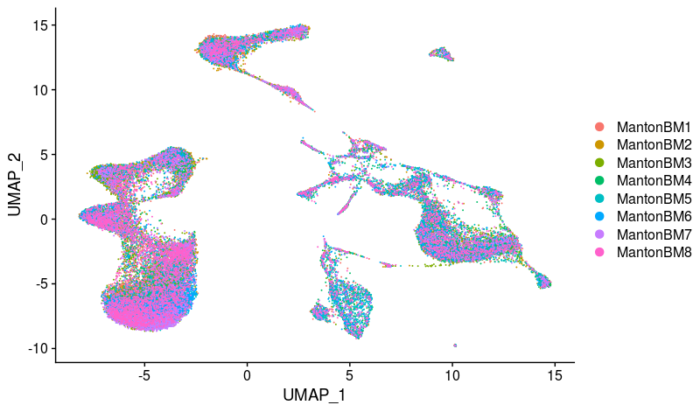
最后贴个完整代码:
- setwd('/your/path/to/seurat')
- #使用reciprocal PCA的方法寻找数据集之间的anchor,从而进行整合
- #数据使用SeuratData提供的人类免疫细胞图谱的数据集的downsample,共40k个细胞
- #没有用完整数据是因为我根据提示找不到原始数据,我好菜
- #加载包
- library(Seurat)
- library(SeuratData)
- library(ggplot2)
- #用于并行计算的包
- library(future)
- library(future.apply)
- #初始化设置
- plan("multiprocess", workers = 4)
- options(future.globals.maxSize = 20000 * 1024^2)
- #加载数据
- # AvailableData()
- # InstallData("hcabm40k.SeuratData")
- data(hcabm40k)
- #将数据按来源拆分,并行计算标准化并寻找差异表达基因各2000个
- bm40k.list <- SplitObject(hcabm40k, split.by = "orig.ident")
- bm40k.list <- lapply(X = bm40k.list, FUN = function(x) {
- x <- NormalizeData(x, verbose = FALSE)
- x <- FindVariableFeatures(x, verbose = FALSE)
- })
- #我猜测在这里运行FindVariableFeatures是为了节省下面scale步骤中的时间,因为scale默认只scale差异表达基因
- #挑选用于整合的基因
- features <- SelectIntegrationFeatures(object.list = bm40k.list)
- #rPCA整合前,先scale再做PCA
- bm40k.list <- lapply(X = bm40k.list, FUN = function(x) {
- x <- ScaleData(x, features = features, verbose = FALSE)
- x <- RunPCA(x, features = features, verbose = FALSE)
- })
- #使用一个男人和一个女人的数据作为reference进行整合,注意降维选择rpca
- anchors <- FindIntegrationAnchors(object.list = bm40k.list, reference = c(1, 2), reduction = "rpca", dims = 1:30)
- bm40k.integrated <- IntegrateData(anchorset = anchors, dims = 1:30)
- #下游分析与可视化
- bm40k.integrated <- ScaleData(bm40k.integrated, verbose = T)
- bm40k.integrated <- RunPCA(bm40k.integrated, verbose = T)
- bm40k.integrated <- RunUMAP(bm40k.integrated, dims = 1:30)
- DimPlot(bm40k.integrated, group.by = "orig.ident")
OK,那么使用seurat进行单细胞RNA-seq数据整合的教程就到这里结束了。