

本来是想出一期使用Monocle3进行单细胞RNA-seq数据分析的教程的,可是Monocle3我死活都装不上,不是依赖报错就是GitHub报错,所以无可奈何只能暂时先使用Monocle2,等后面Monocle3的版本更稳定后再去探索吧。单细胞RNA-seq可以帮助我们在复杂的生物学过程和高度异质的细胞类群中去刻画转录组的变化,进而去研究生物学过程的调控机制或者用特定的基因表达去标记特定的细胞类群或者分化过程中的某些中间态的细胞。在细胞分化发育的过程中都会执行一套特定的基因表达程序,而在整体的生物学过程中,各个细胞执行这套程序往往是不同步的,monocle正是利用了这一特点,将测序得到的细胞放置在计算得到的一条轨迹上,从而刻画出生物学过程(如发育分化等)中细胞的伪时序发展路径,并进一步提供聚类和差异表达分析等手段帮助我们更好的理解生物过程发展和调控的机制。关于更多的算法,可以参考Monocle2的官网资料。
下面总结了Monocle2的主要流程与功能:
- 构建CellDataSet对象,存储单细胞RNA-seq表达矩阵(exprs)与元数据(phenoData和featureData)。
- 用已知的marker基因去区分细胞类型。
- 聚类、区分和计数细胞:发现新的细胞亚型。
- 构建单细胞路径:发现在发育、疾病和生命过程中,细胞从一种状态向另一种状态转变的路径。
- 差异表达分析。
Monocle2既可以使用相对表达水平矩阵(如用Cufflinks计算出的FPKM或TPM),也可以使用绝对的counts数(如UMI表),另外monocle2可以使用10X的分析工具CellRanger处理过的数据直接进行下游分析。但需要注意的是,如果没有UMI,则尽量使用FPKM或TPM而不要使用原始的counts,因为原始的counts数和表达水平之间是存在gap的。OK,那我们结合具体的代码来演示。
CellDataSet类
首先要先来说下CellDataSet类,这个类可以通过给出指定的表达矩阵和元数据进行构建,代码如下。
- #讲解,不要运行
- #导入三组相对应的表达矩阵和元数据
- HSMM_expr_matrix <- read.table("fpkm_matrix.txt")
- HSMM_sample_sheet <- read.delim("cell_sample_sheet.txt")
- HSMM_gene_annotation <- read.delim("gene_annotations.txt")
- #构建monocle中的对象
- pd <- new("AnnotatedDataFrame", data = HSMM_sample_sheet)
- fd <- new("AnnotatedDataFrame", data = HSMM_gene_annotation)
- HSMM <- newCellDataSet(as.matrix(HSMM_expr_matrix),phenoData = pd, featureData = fd)
同样的,CellDataSet也可以与其他的包,如seurat和scater等进行流通,也就是对这三个类进行数据的导入导出,代码如下。
- #讲解,不要运行
- importCDS(data_to_be_imported)
- # 我们可以设置参数import_all来选择是否将所有的seurat和scater对象都进行导入
- #默认import_all为false,或者只保留最小的数据集
- importCDS(data_to_be_imported, import_all = TRUE)
- #当然也可以导出CellDataSet为seurat对象或SCESet
- lung <- load_lung()
- # 导出为seurat对象
- lung_seurat <- exportCDS(lung, 'Seurat')
- # 导出为SCESet
- lung_SCESet <- exportCDS(lung, 'Scater')
另外,很重要的一件事是要为表达矩阵选择一种分布。Monocle2既可以对基于counts数的表达矩阵(如UMI)进行处理,也可以对相对表达矩阵(如FPKM和TPM)进行处理,当然,对UMI的处理效果是最好的。一般来说,对FPKM/TPM进行处理选择对数正态分布,对UMI或counts数进行处理则使用负二项分布。值得注意的是,如果使用relative2abs()函数将FPKM/TPM转换成了counts数,则也可以使用负二项分布进行处理,并且这样的结果往往会比直接使用对数正态分布来的准确。对分布的选择可以通过newCellDataSet函数中的expressionFamily参数进行设定,不同的参数所适用的范围如下截图所示。
- #讲解,请勿运行
- HSMM <- newCellDataSet(count_matrix,phenoData = pd,featureData = fd,expressionFamily=negbinomial.size())

接着呢,单细胞RNA-seq数据量大,并且现有的技术也无法捕捉单个细胞中绝大多数的mRNA,因此使用稀疏矩阵可以大大减少内存占用量和计算时间。
- #讲解,请勿运行
- HSMM <- newCellDataSet(as(umi_matrix, "sparseMatrix"),phenoData = pd,featureData = fd,lowerDetectionLimit = 0.5,expressionFamily = negbinomial.size())
最后,如果使用了10X Genomics数据和cellrangerRkit进行处理,则可直接导入Monocle2进行下游分析。
- #讲解,请勿运行
- cellranger_pipestance_path <- "/path/to/your/CellRange/output/directory"
- gbm <- load_cellranger_matrix(cellranger_pipestance_path)
- fd <- fData(gbm)
- # 下面的colname()[2]中的2是随意选择的,只需要对应于featureData中注释基因缩写的那一列就行
- colnames(fd)[2] <- "gene_short_name"
- gbm_cds <- newCellDataSet(exprs(gbm),phenoData = new("AnnotatedDataFrame", data = pData(gbm)),featureData = new("AnnotatedDataFrame", data = fd),lowerDetectionLimit = 0.5,expressionFamily = negbinomial.size())
创建CellDataSet类
OK,铺垫完CellDataSet相关的一些信息后,就可以结合monocle包提供的HSMM数据进行实战了。首先先加载包和数据,将FPKM/TPM转换成RPC(mRNA per cell),再进行负二项分布处理(这样的好处是往往能得到比直接用对数正态分布处理更精确的数据)。
- setwd('/your/path/to/monocle')
- #导入包
- library(monocle)
- library(HSMMSingleCell)
- data("HSMM_expr_matrix")
- data("HSMM_gene_annotation")
- data("HSMM_sample_sheet")
- #使用相对表达举证先创建一个CellDataSet对象
- pd <- new("AnnotatedDataFrame", data = HSMM_sample_sheet)
- fd <- new("AnnotatedDataFrame", data = HSMM_gene_annotation)
- HSMM <- newCellDataSet(as.matrix(HSMM_expr_matrix),phenoData = pd,featureData = fd,lowerDetectionLimit = 0.1,expressionFamily = tobit(Lower = 0.1))
- #使用CellDataSet估计RNA counts数
- rpc_matrix <- relative2abs(HSMM, method = "num_genes")
- #使用RNA counts数构建一个新的CellDataSet对象
- HSMM <- newCellDataSet(as(as.matrix(rpc_matrix), "sparseMatrix"),phenoData = pd,featureData = fd,lowerDetectionLimit = 0.5,expressionFamily = negbinomial.size())
接下来要估计数据集的大小与离散程度。估计数据集的大小因子可以标准化细胞之间的表达量差异,而数据集的离散程度,则将在后面的差异表达分析中加以运用。需要注意的是,估计数据集大小因子和离散程度需要且只需要在使用负二项分布处理表达矩阵的情况下。
- #估计数据集大小因子和离散程度
- HSMM <- estimateSizeFactors(HSMM)
- HSMM <- estimateDispersions(HSMM)
质控,剔除低质量的数据
接下来就可以进行质控,剔除一些低质量的数据。在单细胞RNA-seq测序过程中,往往会引入一些死细胞或多个细胞黏在一起形成的细胞团,这些数据往往会给下游的分析带来outlier等多种因素的影响,因此需要先进行质控,剔除这些无效的数据。无效的数据主要有两个方面,一是没有多少细胞表达的基因,这些基因对于我们的分析往往没有太大的作用,反而占用大量的计算资源;另一个方面是质量较差的文库,如空细胞,细胞团等情况。因此下面的代码主要从这两个方面进行质控,并筛选出有效的数据。
- > #查看每个基因都有多少细胞表达
- > HSMM <- detectGenes(HSMM, min_expr = 0.1)
- > print(head(fData(HSMM)))
- gene_short_name biotype
- ENSG00000000003.10 TSPAN6 protein_coding
- ENSG00000000005.5 TNMD protein_coding
- ENSG00000000419.8 DPM1 protein_coding
- ENSG00000000457.8 SCYL3 protein_coding
- ENSG00000000460.12 C1orf112 protein_coding
- ENSG00000000938.8 FGR protein_coding
- num_cells_expressed use_for_ordering
- ENSG00000000003.10 184 FALSE
- ENSG00000000005.5 0 FALSE
- ENSG00000000419.8 211 FALSE
- ENSG00000000457.8 18 FALSE
- ENSG00000000460.12 47 TRUE
- ENSG00000000938.8 0 FALSE
- #######################################################
对基因进行筛选,选出的基因至少在10个细胞中有表达。
- #以10个细胞为阈值进行筛选
- expressed_genes <- row.names(subset(fData(HSMM),num_cells_expressed >= 10))
接下来对细胞进行筛选,对细胞的筛选则没有比较统一的标准去判断是否测的足够深,但经验上一般认为,如果测得的reads数小于几千个,那么这个细胞一般不能提供特别有意义的数据。另外,使用FastQC也可以对单细胞状态和测序结果进行额外的筛选。细胞状态的元数据存储在phenoData中,可以使用pData()函数进行调用。
- > print(head(pData(HSMM)))
- Library Well Hours Media Mapped.Fragments Pseudotime
- T0_CT_A01 SCC10013_A01 A01 0 GM 1958074 23.916673
- T0_CT_A03 SCC10013_A03 A03 0 GM 1930722 9.022265
- T0_CT_A05 SCC10013_A05 A05 0 GM 1452623 7.546608
- T0_CT_A06 SCC10013_A06 A06 0 GM 2566325 21.463948
- T0_CT_A07 SCC10013_A07 A07 0 GM 2383438 11.299806
- T0_CT_A08 SCC10013_A08 A08 0 GM 1472238 67.436042
- State Size_Factor num_genes_expressed
- T0_CT_A01 1 1.392811 6850
- T0_CT_A03 1 1.311607 6947
- T0_CT_A05 1 1.218922 7019
- T0_CT_A06 1 1.013981 5560
- T0_CT_A07 1 1.085580 5998
- T0_CT_A08 2 1.099878 6055
如果参照本案例使用了RPC值来表征表达水平,则最好去看一下mRNA的总量在细胞之间的变化。
- #统计各个细胞中mRNA的总表达水平
- pData(HSMM)$Total_mRNAs <- Matrix::colSums(exprs(HSMM))
- HSMM <- HSMM[,pData(HSMM)$Total_mRNAs < 1e6]
- qplot(Total_mRNAs, data = pData(HSMM), color = Hours, geom ="density")
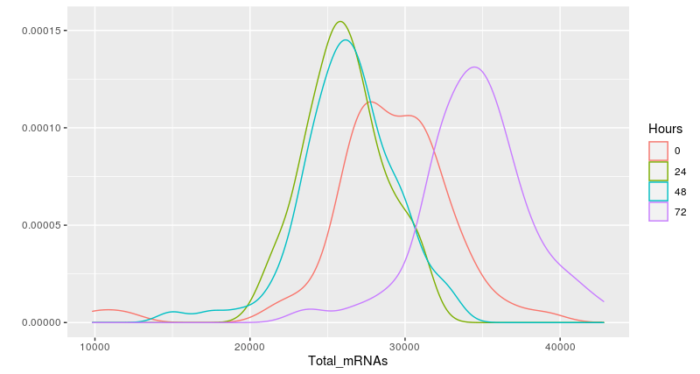
这样就统计出了不同的处理条件下,细胞中mRNA总量的分布图,根据分布图去选择细胞中表达的总mRNA的上下限,这里选择了两倍的标准差。
- #以两倍标准差作为上下限的阈值
- upper_bound <- 10^(mean(log10(pData(HSMM)$Total_mRNAs)) 2*sd(log10(pData(HSMM)$Total_mRNAs)))
- lower_bound <- 10^(mean(log10(pData(HSMM)$Total_mRNAs)) - 2*sd(log10(pData(HSMM)$Total_mRNAs)))
- qplot(Total_mRNAs, data = pData(HSMM), color = Hours, geom ="density")
- geom_vline(xintercept = lower_bound)
- geom_vline(xintercept = upper_bound)
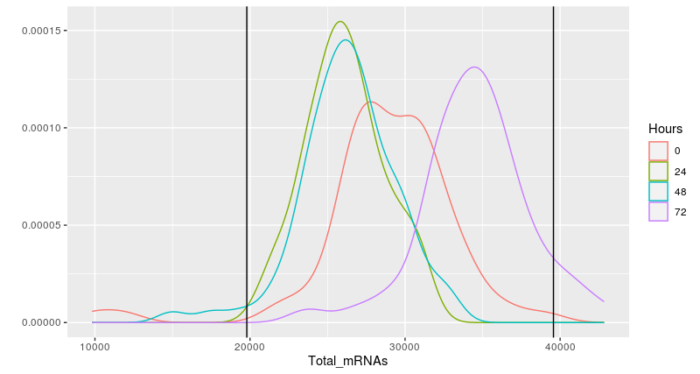
- #筛选,并注意由于剔除了部分细胞,因此基因的相关数据要重新计算
- HSMM <- HSMM[,pData(HSMM)$Total_mRNAs > lower_bound & pData(HSMM)$Total_mRNAs < upper_bound]
- HSMM <- detectGenes(HSMM, min_expr = 0.1)
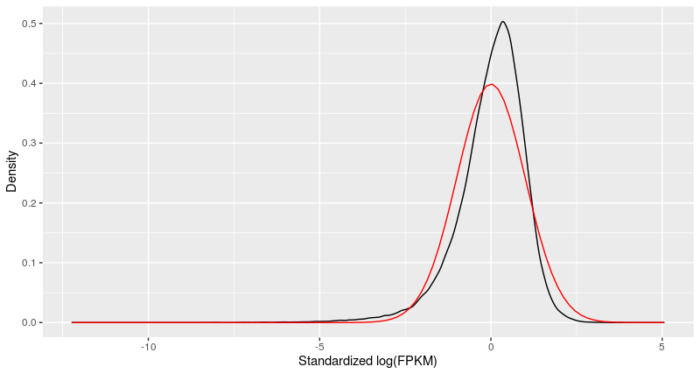
验证筛选细胞后,细胞的mRNA表达水平是否呈现对数正态分布。
- #验证筛选过后,细胞的mRNA表达水平是否为对数正态分布
- #对mRNA表达水平取log
- L <- log(exprs(HSMM[expressed_genes,]))
- #对每个基因进行标准化,并使用melt函数改变数据形式
- melted_dens_df <- melt(Matrix::t(scale(Matrix::t(L))))
- #作图
- qplot(value, geom = "density", data = melted_dens_df)
- stat_function(fun = dnorm, size = 0.5, color = 'red')
- xlab("Standardized log(FPKM)")
- ylab("Density")
鉴定细胞类型
质控完后就可以鉴别细胞类型了。鉴定细胞类型也很容易,只需要用特定的marker基因的表达去进行分选即可,我觉得更困难的点可能在于marker基因的表达阈值的问题,很多细胞都会本底表达一些基因,并且不同的测序方法和批次对表达量的表征都会有差别。这本案例中,我们做了成肌细胞的单细胞RNA-seq,他们通常大量表达MYF5这样的关键基因,而成纤维细胞则高表达ANPEP这样的成纤维细胞特异的基因,因此可以通过这两个基因的表达来区分这两群细胞。Monocle2中使用CellTypeHierarchy类来筛选和存储不同的细胞类型,通过classifyCells函数在CellDataSet对象的phenoData中加入新的一列:CellType来给不同的细胞类型打上标签。
- #鉴定细胞类型
- MYF5_id <- row.names(subset(fData(HSMM), gene_short_name == "MYF5"))
- ANPEP_id <- row.names(subset(fData(HSMM),gene_short_name == "ANPEP"))
- #构建cth类,确定筛选规则
- cth <- newCellTypeHierarchy()
- cth <- addCellType(cth, "Myoblast", classify_func = function(x) { x[MYF5_id,] >= 1 })
- cth <- addCellType(cth, "Fibroblast", classify_func = function(x) { x[MYF5_id,] < 1 & x[ANPEP_id,] > 1 })
- #通过classifyCells函数给phenoData加入新的一列:CellType
- HSMM <- classifyCells(HSMM, cth, 0.1)
这里提一下classifyCells函数中的参数,0.1表示对phenoData中的Cluster列进行分组,如果某一组中的细胞数量超过10%符合某一判断标准,则将这一组全部归于这一类细胞,如果这一组细胞,同时有10%符合一判断标准,又有10%符合另一判断标准,则将这组细胞归于‘Ambiguous’。同样的,如果细胞同时不满足任何一个判断函数,则被归为‘Unknown’。我们来看下上面的分类结果。
- > #查看筛选情况
- > table(pData(HSMM)$CellType)
- Fibroblast Myoblast Unknown
- 56 85 121
用pie图可视化。
- #pie图可视化
- pie <- ggplot(pData(HSMM), aes(x = factor(1), fill = factor(CellType))) geom_bar(width = 1)
- pie coord_polar(theta = "y") theme(axis.title.x = element_blank(), axis.title.y = element_blank())
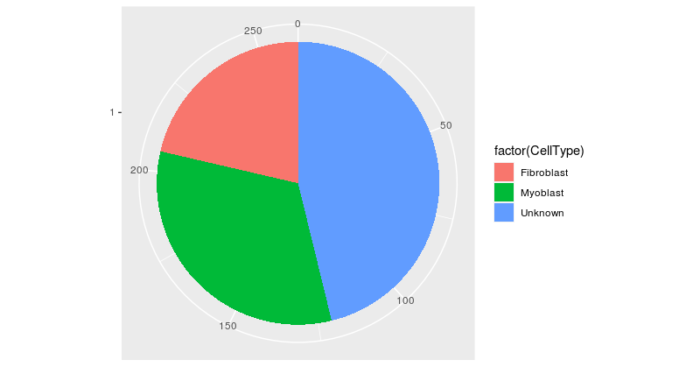
可以看到,我们有将近一半的细胞无法为其赋予类型标签,这大大减少了我们可以进行下游分析的数据量。Monocle2提供了一定的算法来进一步划分这些细胞,其原理是通过clusterCells函数,利用整体的表达谱特征将细胞聚类在一起,这样这些Unknown的细胞即使不高表达MYF5,也能通过整体的表达谱特征将其归为成肌细胞。clusterCells函数能在无监督或半监督(辅以先验知识)的方式进行运算。
利用聚类算法进一步区分细胞类型
无监督算法
我们首先来介绍通过无监督的方式对细胞进行聚类。这里需要考虑用哪些基因进行聚类,如果选择全基因,势必会引入大量的噪声和无意义的计算,因此我们应该基于一定的表达量去选择那些在细胞中差异最大的基因去进行聚类,这样才能非常好的反映出细胞类型的信息。
- #无监督的方法对细胞进行聚类
- #筛选出有一定表达量的基因
- disp_table <- dispersionTable(HSMM)
- unsup_clustering_genes <- subset(disp_table, mean_expression >= 0.1)
- HSMM <- setOrderingFilter(HSMM, unsup_clustering_genes$gene_id)
- plot_ordering_genes(HSMM)
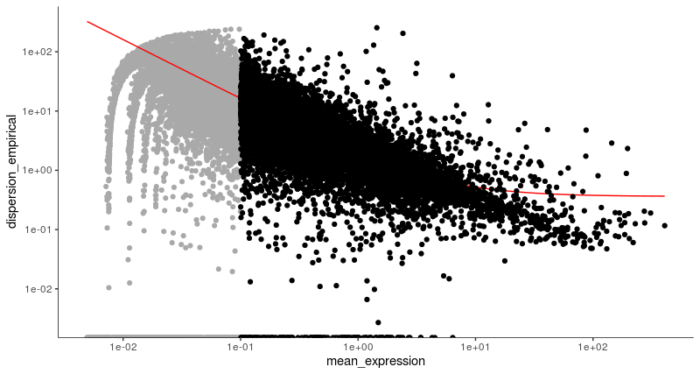
setOrderingFilter函数标记出了我们将用于下游聚类的基因,红线则描述了基于散点图刻画的基因平均表达量和离散程度的预期关系。其中黑色的点表示将用于下游聚类的基因,而灰色的点则表示被舍弃的基因。和seurat类似,Monocle2也使用t-SNE的方法进行降维和聚类,具体t-SNE降维的过程可以参考我之前关于使用seurat进行单细胞RNA-seq聚类分析的文章。
- #t-SNE降维
- #线性降维选择PCA,分析不同的PC轴对变异的贡献,默认选择log函数进行标准化
- plot_pc_variance_explained(HSMM, return_all = F)
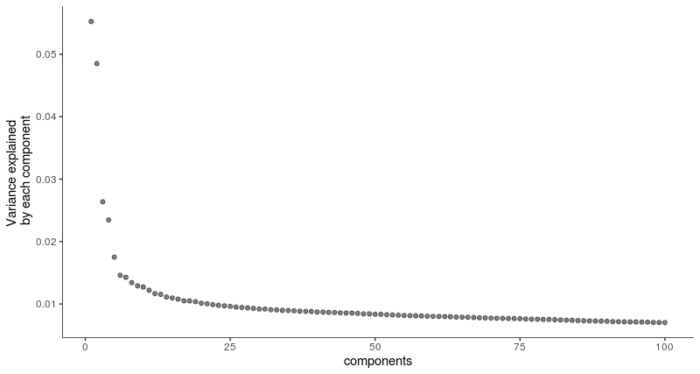
在做PCA主成分分析时默认选择log函数对表达量进行标准化,选择前6个PC进行下一步降维聚类和可视化。
- #选择前6个PC进行降维聚类
- HSMM <- reduceDimension(HSMM, max_components = 2, num_dim = 6, reduction_method = 'tSNE', verbose = T)
- HSMM <- clusterCells(HSMM, num_clusters = 2)
- plot_cell_clusters(HSMM, 1, 2, color = "CellType")
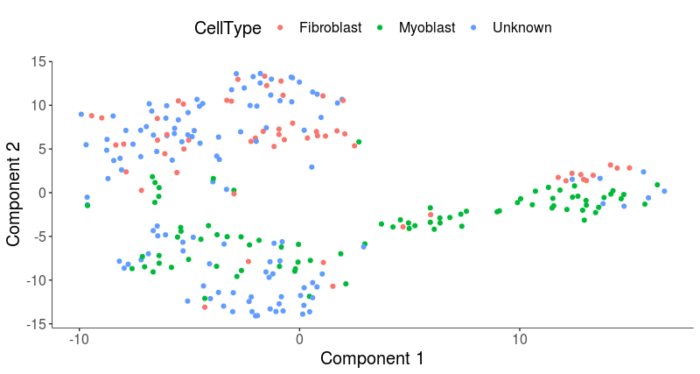
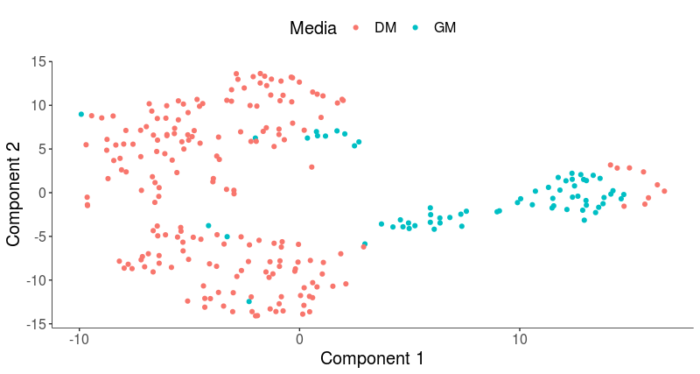
可以看到,成肌细胞和成纤维细胞并没有在降维图上很好地区分成两群,当然,降维和聚类并不能保证就一定能得到我们想要的生物学差异。但在本案例中,除了细胞类型外,还存在其他的导致细胞表达谱变化的差异来源。为了诱导肌细胞分化,我们需要将细胞从GM培养基转移到DM培养基,因此培养条件也是差异的一个来源,可以在phenoData中找到Media的标签。
- #培养基引入的差异
- plot_cell_clusters(HSMM, 1, 2, color = "Media")
Monocle2允许我们使用residualModelFormulaStr参数从差异中减去我们认为无意义的差异,从而得到更符合我们的生物学问题的聚类结果,这里我们减去了培养基和细胞内基因表达量所引入的差异。
- #减去培养基和基因表达量所引入的差异
- HSMM <- reduceDimension(HSMM, max_components = 2, num_dim = 2, reduction_method = 'tSNE', residualModelFormulaStr = "~Media num_genes_expressed", verbose = T)
- HSMM <- clusterCells(HSMM, num_clusters = 2)
- plot_cell_clusters(HSMM, 1, 2, color = "CellType")
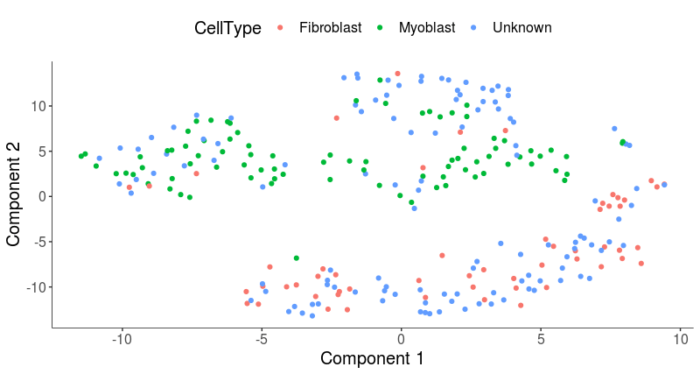
可以看到,会比之前的聚类结果好一些,但也没有达到非常完美的程度,下面我们具体从三中细胞类型来看聚类的情况。
- #从三种细胞类型来看聚类情况
- HSMM <- clusterCells(HSMM, num_clusters = 2 )
- plot_cell_clusters(HSMM, 1, 2, color = "Cluster") facet_wrap(~CellType)
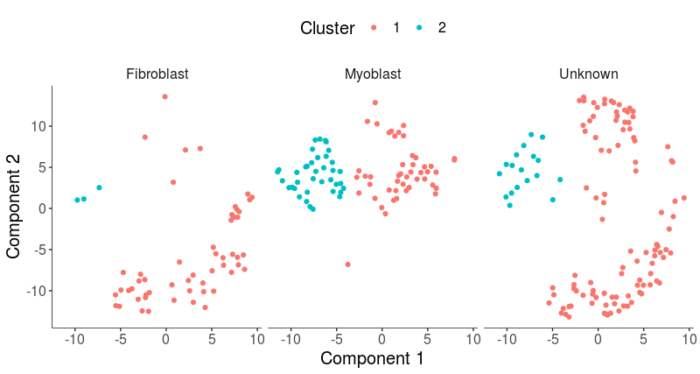
可以看到成纤维细胞明显被聚到了cluster1,而成肌细胞的聚类结果不是很理想,并没能被单独聚到cluster2中,因此这样的聚类结果,实际上并不能非常完美地表征成纤维细胞和成肌细胞之间的差异,对于unknown细胞的分类效果,也并没有想象中的那么理想。造成这样的结果,一方面可能是marker基因的特异性存疑,另一方面可能是算法的问题,为了解决后者,我们接下来尝试使用先验知识的半监督聚类。
半监督算法
在半监督的聚类过程中,我们将使用先验知识(marker基因)先挑选出在细胞群体内部和marker基因一同高差异表达的基因作为list,检验这些基因对两类细胞表达的特异性,从而获得一个合适的基因子集来进行后续的降维聚类。
- #半监督的方法对基因进行聚类
- #去除培养基和基因数的差异,计算出和marker基因共同差异表达的基因
- marker_diff <- markerDiffTable(HSMM[expressed_genes,], cth, residualModelFormulaStr = "~Media num_genes_expressed", cores = 1)
markerDiffTable函数会把CellTypeSet对象和CellTypeHierarchy对象吃掉,筛选出对应的两群细胞,并把Unknown和Ambiguous的细胞剔除,在剩下的细胞中寻找差异表达的基因。当然,我们这里也去除了培养基和基因数所引入的差异。得到了这样一组基因list后,进一步对其差异表达的显著性和细胞类型的特异性进行筛选。
- #对显著性和细胞类型的特异性进行筛选
- candidate_clustering_genes <- row.names(subset(marker_diff, qval < 0.01))
- marker_spec <- calculateMarkerSpecificity(HSMM[candidate_clustering_genes,], cth)
- head(selectTopMarkers(marker_spec, 3))
从而,我们得到了各个基因所存在的细胞类型及其细胞类型特异性打分,这种特异性的打分可以参考文章Cabili et al,简单来说特异性打分从0到1,越接近1,则这种基因越局限于在对应地细胞类型中表达。为每种细胞类型挑选最特异的500个基因用于后续的降维聚类。
- #为每种细胞类型挑选最特异的500个基因用于后续的降维聚类
- semisup_clustering_genes <- unique(selectTopMarkers(marker_spec, 500)$gene_id)
- HSMM <- setOrderingFilter(HSMM, semisup_clustering_genes)
- plot_ordering_genes(HSMM)
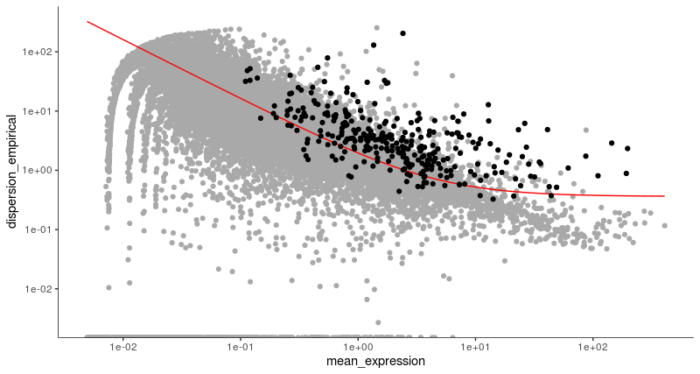
可以看到这些基因相对来说表达水平较高,在细胞之间的离散程度也较大,符合我们选择差异基因的条件。接下来的步骤就和之前的聚类降维类似。
- #t-SNE降维并聚类
- #PC轴对差异的贡献
- plot_pc_variance_explained(HSMM, return_all = F)
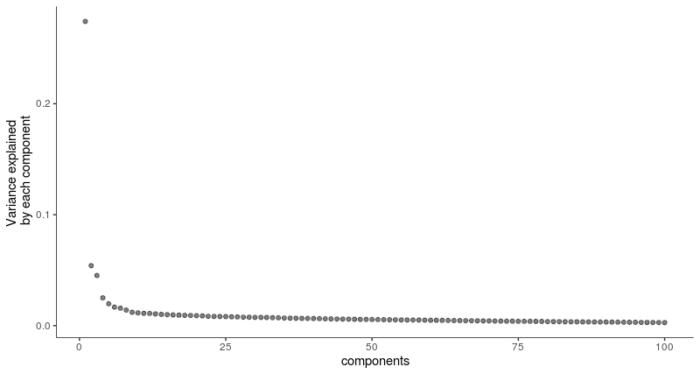
可以看到前3个PC轴就已经能解释绝大多数的差异,因此后续的t-SNE降维,我们就选择前三个PC作为高维向量。
- #t-SNE
- HSMM <- reduceDimension(HSMM, max_components = 2, num_dim = 3, norm_method = 'log', reduction_method = 'tSNE', residualModelFormulaStr = "~Media num_genes_expressed", verbose = T)
- HSMM <- clusterCells(HSMM, num_clusters = 2)
- plot_cell_clusters(HSMM, 1, 2, color = "CellType")
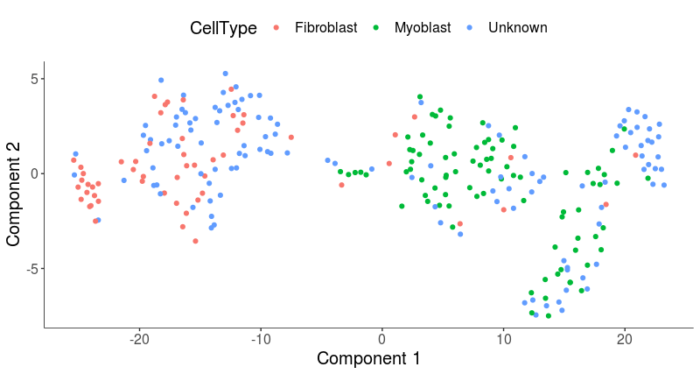
在降维图上,成纤维细胞和成肌细胞已经区分得比较开了,我们再看下聚类的情况是否能把成纤维细胞和成肌细胞更好地区分开。
- #从三种细胞类型来看聚类情况
- HSMM <- clusterCells(HSMM, num_clusters = 2)
- plot_cell_clusters(HSMM, 1, 2, color = "Cluster") facet_wrap(~CellType)
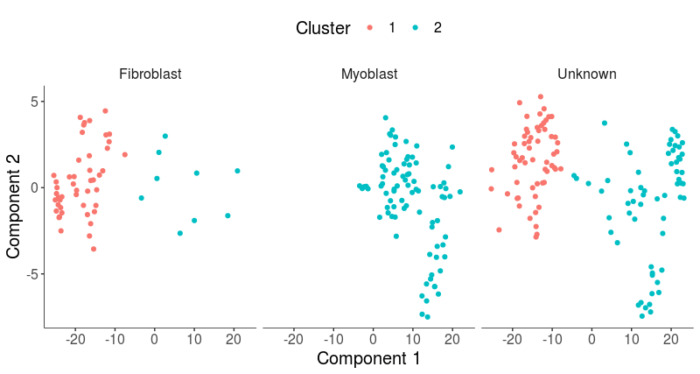
可以看到cluster1对应于成纤维细胞,cluster2对应于成肌细胞,相对于无监督的聚类,通过cluster区分细胞类型的准确率在成纤维细胞类型中略有降低,但在成肌细胞类型中显著升高。现在我们就可以把unknown的细胞依据cluster分配到两种细胞类型,其原理与上面所说的classifyCells函数类似。以0.1举例,如果某一cluster中的细胞有10%以上符合某一判断标准,则把这一cluster的细胞全部标记为这一类型的细胞,如果某一组cluster中的细胞既有10%的细胞符合某一判断标准,又有10%的细胞符合另一判断标准,则把这一cluster的细胞标记为‘Ambiguous’。通过这样的算法,就可以将之前的Unknown的细胞,通过聚类给他们贴上相应的细胞类型标签,尽管他们可能不满足单一的marker基因的判断标准。
- #将Unknown的细胞进行划分
- HSMM <- clusterCells(HSMM, num_clusters = 2, frequency_thresh = 0.1, cell_type_hierarchy = cth)
- plot_cell_clusters(HSMM, 1, 2, color = "CellType")
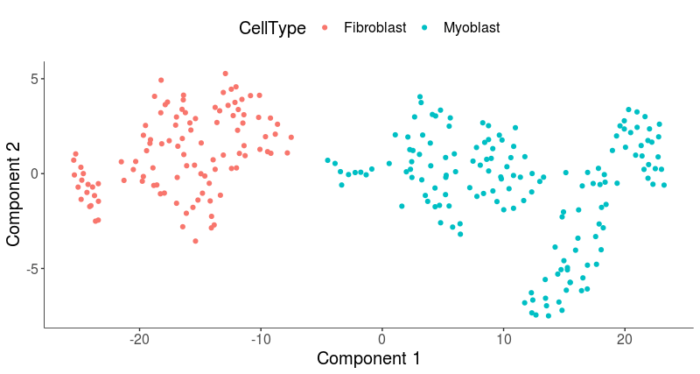
这样就通过cluster的方法,将Unknown的细胞也分配到了两种细胞类型中,最后看下两种细胞的比例。
- #看下两种细胞的比例
- pie <- ggplot(pData(HSMM), aes(x = factor(1), fill = factor(CellType)))
- geom_bar(width = 1)
- pie
- coord_polar(theta = "y")
- theme(axis.title.x = element_blank(), axis.title.y = element_blank())
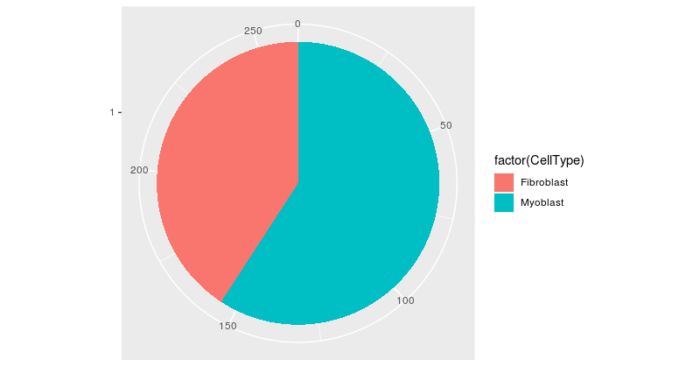
我们接下来的分析就只针对成肌细胞,将成纤维细胞全部剔除掉。
- #仅取成肌细胞用作后续的分析
- HSMM_myo <- HSMM[,pData(HSMM)$CellType == 'Myoblast']
构建单细胞路径
筛选出我们想要特别研究的细胞类型后,我们就可以构建单细胞路径,进行伪时序分析了。这里我们要先讲一讲对伪时序的理解。细胞在自然状态下,接受相应的刺激或仅仅随着时间的流逝,细胞的状态会发生相应的改变,这种改变体现在一部分基因的沉默和一部分基因的表达上。这种转变如何发生是我们感兴趣的问题,但同样面临着一些困难:在不杀死细胞的前提下,我们很难追踪一个细胞内完整转录组的变化,并且我们也很难纯化出中间态的细胞进行转录组分析。单细胞RNA-seq为我们克服这些困难提供了可能,单细胞RNA-seq捕捉到了各个细胞的转录组特征,这些细胞由于发展的不同步性,有的处于发展早期,有的处于发展晚期,如果我们能将这些细胞放置在一定的路径的合适位置上,我们就能刻画出某一生物学事件中细胞变化的过程,也就描绘出了细胞状态发生改变的‘伪’时序,这种伪时序同样能让我们窥探细胞状态转变过程中转录组的变化,一旦构建好了单细胞路径,就可以使用Monocle提供的差异表达分析工具去探索这一路径上的基因表达调控。如果这种状态转变最终出现多种细胞类型,则意味着在单细胞路径中存在分支,这种分支的产生和发展对应了细胞命运的决定,也是值得我们探讨的问题。Monocle2使用一种称为‘reversed graph embedding ’的机器学习方法学习和构建单细胞路径,具体的算法原理可以参考官方的说明文档。构建单细胞路径的流程主要分为以下三个部分。
- 挑选用于定义生物学过程的基因(feature selection):单细胞RNA-seq数据量大,噪声高,需要对基因进行一定的筛选,又不能遗漏那些提供细胞状态信息的重要基因。和之前挑选基因用于降维聚类类似,提供无监督和半监督的算法。
- 降维:使用‘reversed graph embedding ’的机器学习算法对数据进行降维。
- 用伪时间排列细胞:把表达谱数据投影到低维空间后,Monocle就可以学习描述细胞从一种状态转移到另一种状态的路径了。Monocle假定这种路径具有树形结构,即开始转变的细胞在根处,沿着路径和分叉(如果有的话),最终到达叶处,也就是转变的最终状态。一个细胞的伪时值就是该细胞位置离根的距离。
使用不同状态间差异表达基因构建单细胞路径
首先我们选择用于后续构建单细胞路径的基因,这些基因应当在肌肉发育的过程中呈现增加或下降的趋势。我们首先先尝试用无监督的算法来进行挑选,这种算法的想法也很简单,既然这些基因在生物学过程中会出现逐渐的下降或增加,那我直接将首尾两种细胞的差异表达基因找出来作为我的基因列表即可。
- #构建单细胞路径
- #挑选用于后续构建路径的基因
- #无监督的算法,首尾两种细胞的差异表达基因
- diff_test_res <- differentialGeneTest(HSMM_myo[expressed_genes,], fullModelFormulaStr = "~Media")
- ordering_genes <- row.names (subset(diff_test_res, qval < 0.01))
- HSMM_myo <- setOrderingFilter(HSMM_myo, ordering_genes)
- plot_ordering_genes(HSMM_myo)
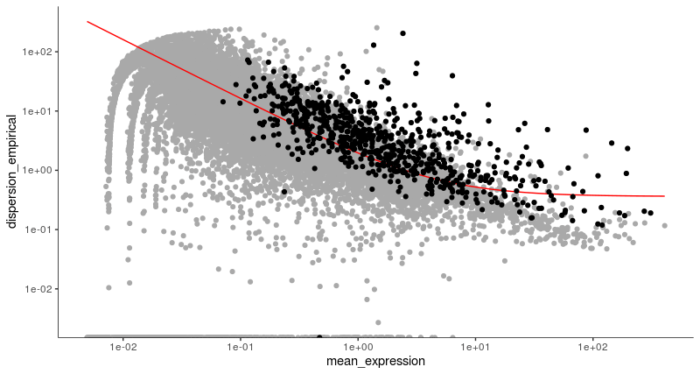
第二步进行降维,使得最终的路径便于可视化和理解。
- #降维
- HSMM_myo <- reduceDimension(HSMM_myo, max_components = 2, method = 'DDRTree')
第三步将降维后的数据填充到路径的相应位置上,并可视化。
- #构建路径并可视化
- HSMM_myo <- orderCells(HSMM_myo)
- plot_cell_trajectory(HSMM_myo, color_by = "Hours")
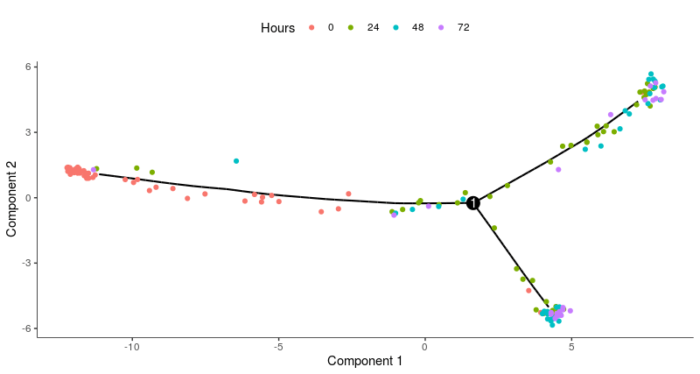
可以看到,可视化后得到典型的树形结构,0小时的细胞主要集中在最左侧,72小时的细胞则主要集中在最右侧的两个分支上。但Monocle2并不能先天知道哪个状态是根,哪个状态是叶,因此需要在orderCells函数中设置root_state参数指定某一state的细胞作为根,state是Monocle2对树形结构的拆解所划分出的不同的状态,可以看看树形结构的不同位置分别被归为哪一种state。
- #树形结构state
- plot_cell_trajectory(HSMM_myo, color_by = "State")
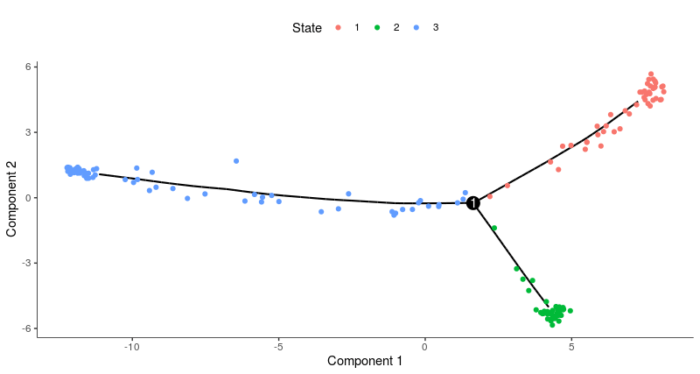
下面我们使用简单的函数去判断那种state的细胞含有更多0小时的细胞,那么这种state所在的位置就被定义为根,从而我们便可以得到细胞的伪时值。
- #判断那种state含有更多的0小时的细胞从而确定根
- GM_state <- function(cds){
- if (length(unique(pData(cds)$State)) > 1){
- T0_counts <- table(pData(cds)$State, pData(cds)$Hours)[,"0"]
- return(as.numeric(names(T0_counts)[which(T0_counts == max(T0_counts))]))
- } else {
- return (1)
- }
- }
- HSMM_myo <- orderCells(HSMM_myo, root_state = GM_state(HSMM_myo))
- plot_cell_trajectory(HSMM_myo, color_by = "Pseudotime")
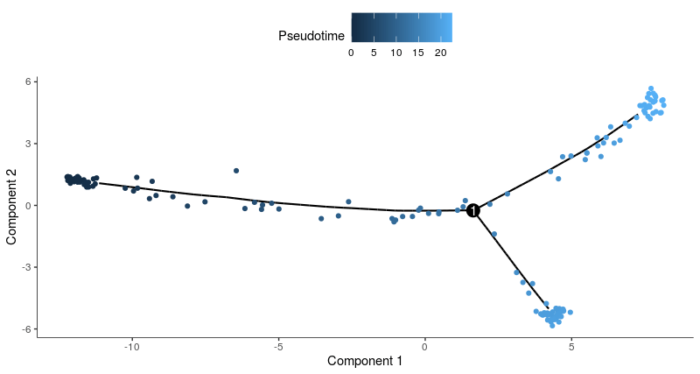
当然,如果树形结构中的state太多,也可以使用facet分页显示不同的state。
- #分页显示不同的state
- plot_cell_trajectory(HSMM_myo, color_by = "State")
- facet_wrap(~State, nrow = 1)
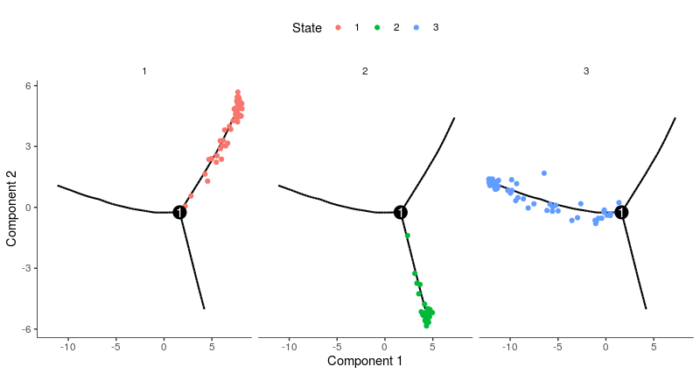
另外,如果实验设计的时候没有时间序列,则需要通过marker基因来确定哪个state的细胞处于根的位置。以这个实验为例,处于根的细胞应当更多的表达增殖的基因,而位于叶的基因则应当表达MyoD等终末分化基因。
- #从marker基因的表达来确定哪个state为根
- blast_genes <- row.names(subset(fData(HSMM_myo), gene_short_name %in% c("CCNB2", "MYOD1", "MYOG")))
- plot_genes_jitter(HSMM_myo[blast_genes,], grouping = "State", min_expr = 0.1)
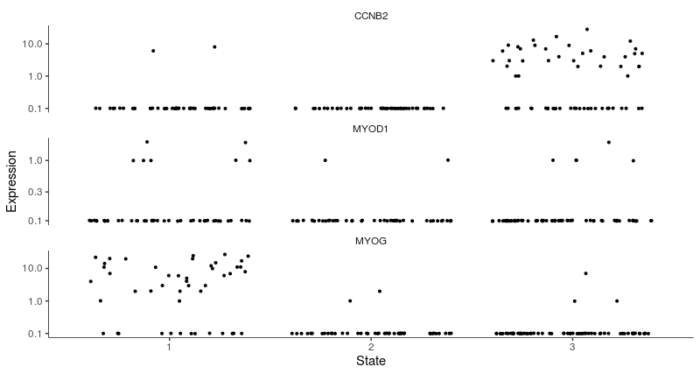
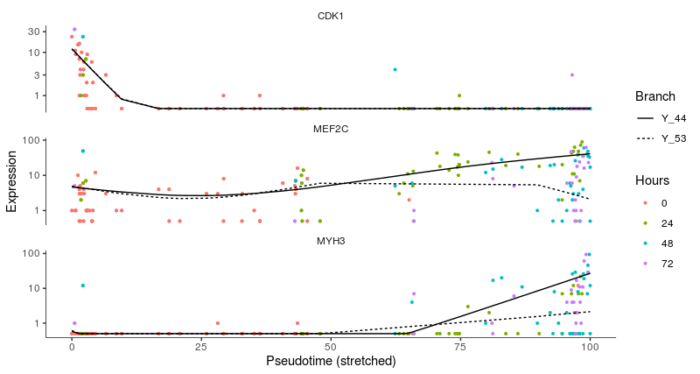
可以看到CCNB2在state3中高表达,而MyoD和MyoG在state1中高表达,与上面分析所得的的伪时序是一致的。为了进一步验证,我们可以挑选一些肌肉发育过程中的marker基因,观察其沿着伪时序轴的变化规律。
- #marker基因随伪时序的变化规律,进一步验证可靠性
- HSMM_expressed_genes <- row.names(subset(fData(HSMM_myo), num_cells_expressed >= 10))
- HSMM_filtered <- HSMM_myo[HSMM_expressed_genes,]
- my_genes <- row.names(subset(fData(HSMM_filtered), gene_short_name %in% c("CDK1", "MEF2C", "MYH3")))
- cds_subset <- HSMM_filtered[my_genes,]
- plot_genes_in_pseudotime(cds_subset, color_by = "Hours")
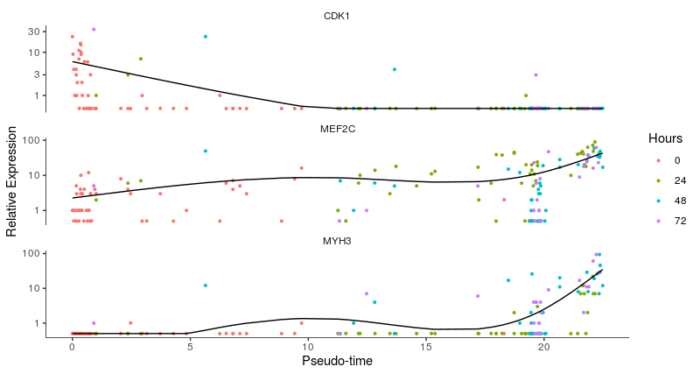
这里最后提一下,如果Monocle2所构建的单细胞路径没有分支,则只会有一种state,因此无法用上述的函数确定root,在这种情况下可以使用orderCells中的reverse参数,对根和叶进行倒置,最终调整到合适的状态。上面的内容都是基于无监督的算法挑选下游分析的基因所得到的结果,下面我们再来谈谈其他用于挑选下游分析的基因的算法。
- ‘dqFeature’法,官网推荐的无监督算法,基于聚类的结果寻找差异表达基因。
- 使用基因表达在细胞间的离散程度来进行选择,方差越大,这样的基因往往能提供更多的信息。
- 半监督算法,基于marker基因进行挑选,原理和降维聚类前所使用的半监督算法挑选基因是类似的。
dqFeature法挑选基因构建单细胞路径
首先我们来尝试‘dpFeature’法,这也是官网最推荐的无监督算法。其原理是先将数据进行降维(t-SNE),在二维的降维图上,使用密度峰值算法(densityPeak)进行聚类,这种算法的想法也很简单,先计算出每个细胞周围的细胞密度P和该细胞离周围细胞的最小距离Δ,通过一定的阈值选择,最终划分出不同数量的cluster。最后挑选出cluster之间的差异表达基因由于构建单细胞路径。
- #其他的挑选用于构建单细胞路径的基因的方法
- #dqFeature法
- #t-SNE降维
- #先挑选出至少在5%的细胞中有表达的基因
- HSMM_myo <- detectGenes(HSMM_myo, min_expr = 0.1)
- fData(HSMM_myo)$use_for_ordering <- fData(HSMM_myo)$num_cells_expressed > 0.05 * ncol(HSMM_myo)
先挑选出至少在5%的细胞中都有表达的基因,再进行PCA主成分分析,挑选用于后续降维的PC轴。
- #PCA主成分分析
- plot_pc_variance_explained(HSMM_myo, return_all = F)
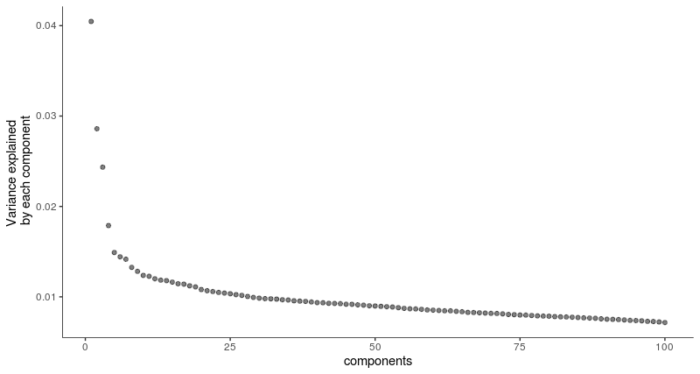
选择前4个PC用于t-SNE降维聚类。
- #选择前4个PC用于t-SNE降维
- HSMM_myo <- reduceDimension(HSMM_myo, max_components = 2, norm_method = 'log', num_dim = 4, reduction_method = 'tSNE', verbose = T)
- #默认参数聚类
- HSMM_myo <- clusterCells(HSMM_myo, verbose = F)
- #可视化
- plot_cell_clusters(HSMM_myo, color_by = 'as.factor(Cluster)')
- plot_cell_clusters(HSMM_myo, color_by = 'as.factor(Hours)')
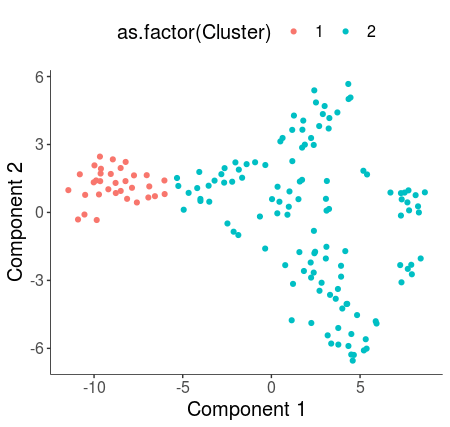
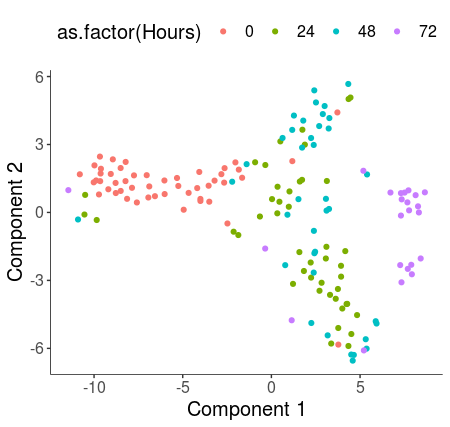
可以看到使用默认参数只聚出了两类,但和Hours参数所显示出的pattern很相似,因此我们可以通过优化参数以使得聚类的结果和Hours参数所表征的类更为接近。
- #调整参数
- plot_rho_delta(HSMM_myo)
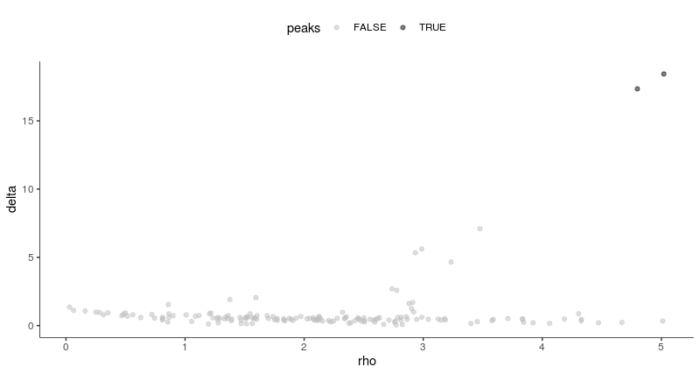
这张图表征的是使用默认参数的情况下所聚出的类(图中的黑点),通过调整阈值,我们可以调整我们所聚出的类,或者直接使用num_clusters参数进行调整。这里选择delta阈值为4,rho阈值为3。
- #选择delta阈值为4,rho阈值为3
- plot_rho_delta(HSMM_myo, rho_threshold = 3, delta_threshold = 4)
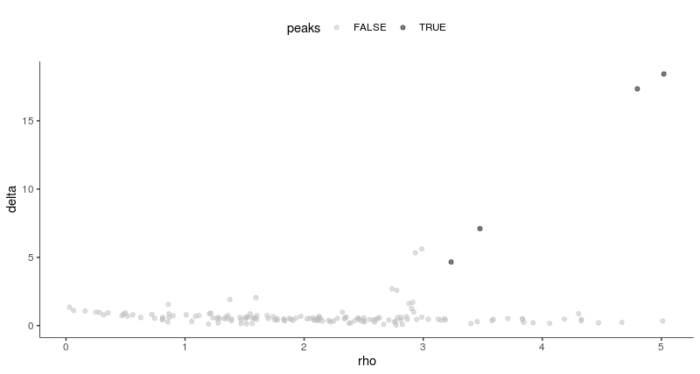
通过调整参数,我们便将cluster的数量调整到了4,接下来可视化看下与Hours参数所表征的细胞类群的相似程度,这里需要将skip_rho_sigma参数设置为TRUE。
- #聚类,可视化
- HSMM_myo <- clusterCells(HSMM_myo, rho_threshold = 3, delta_threshold = 4, skip_rho_sigma = T, verbose = F)
- plot_cell_clusters(HSMM_myo, color_by = 'as.factor(Cluster)')
- plot_cell_clusters(HSMM_myo, color_by = 'as.factor(Hours)')
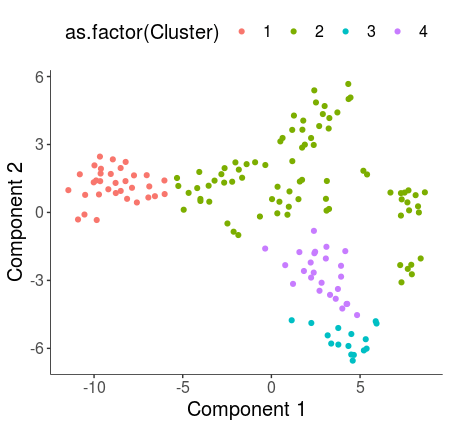
显然参数选择并不好,尝试delta等于5,rho等于2。
- #选择delta阈值为5,rho阈值为2
- HSMM_myo <- clusterCells(HSMM_myo, rho_threshold = 2, delta_threshold = 5, skip_rho_sigma = T, verbose = F)
- plot_cell_clusters(HSMM_myo, color_by = 'as.factor(Cluster)')
- plot_cell_clusters(HSMM_myo, color_by = 'as.factor(Hours)')
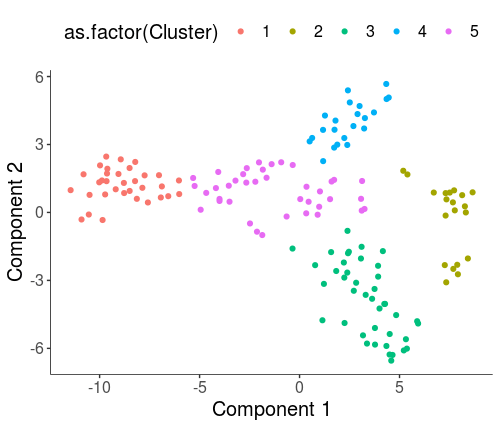
这样的参数选择,就使得cluster更能反映真实的生物学状态了。接下来就是计算cluster之间的差异表达基因了。
- #计算cluster之间的差异表达基因
- clustering_DEG_genes <- differentialGeneTest(HSMM_myo[HSMM_expressed_genes,], fullModelFormulaStr = '~Cluster', cores = 1)
选择前1000个最显著的差异表达基因用于构建单细胞路径。
- #选择前1000个最显著的差异表达基因用于构建单细胞路径
- HSMM_ordering_genes <- row.names(clustering_DEG_genes)[order(clustering_DEG_genes$qval)][1:1000]
- HSMM_myo <- setOrderingFilter(HSMM_myo, ordering_genes = HSMM_ordering_genes)
- HSMM_myo <- reduceDimension(HSMM_myo, method = 'DDRTree')
- HSMM_myo <- orderCells(HSMM_myo)
- HSMM_myo <- orderCells(HSMM_myo, root_state = GM_state(HSMM_myo))
- plot_cell_trajectory(HSMM_myo, color_by = "Hours")
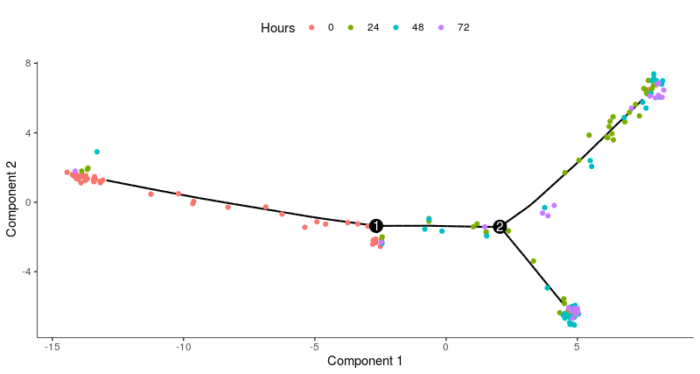
可以看到整体上是类似的,细节上略有区别,可以忽略。
利用基因在细胞间表达的离散程度挑选基因构建单细胞路径
利用基因表达在细胞间的离散程度来进行筛选的想法也很简单,基因表达的差异越大,往往能携带更多的信息,但也要注意的是,方差往往是与表达的均值相关的,因此通过离散程度筛选基因时,也需要考虑表达量的均值。
- #展示,勿运行
- disp_table <- dispersionTable(HSMM_myo)
- ordering_genes <- subset(disp_table, mean_expression >= 0.5 & dispersion_empirical >= 1 * dispersion_fit)$gene_id
半监督算法挑选基因构建单细胞路径
非监督的算法能够很好的排除bias的干扰,但他同样也可能过于专注在一些较大的变化上,而这些变化可能与我们的研究目的没有太大的关系。这就引出了半监督的算法,它能够帮助我们更好地专注于我们感兴趣的问题上,当然,缺点是会引入bias。这种半监督的算法,基于marker基因进行挑选,原理和降维聚类前所使用的半监督算法挑选基因是类似的,同样是找到和marker共同差异表达的基因进行后续分析。在本例肌肉的发育中,早期的细胞应当具有一定的分类增殖能力,而晚期的肌肉细胞则更倾向于表达与肌肉收缩相关的基因。这里选择CCNB2作为早期成肌细胞的marker,MYH3作为晚期肌管细胞的marker。
- #半监督算法,CCNB2作为早期成肌细胞的marker,MYH3作为晚期肌管细胞的marker
- CCNB2_id <- row.names(subset(fData(HSMM_myo), gene_short_name == "CCNB2"))
- MYH3_id <- row.names(subset(fData(HSMM_myo), gene_short_name == "MYH3"))
- cth <- newCellTypeHierarchy()
- cth <- addCellType(cth, "Cycling myoblast", classify_func = function(x) { x[CCNB2_id,] >= 1 })
- cth <- addCellType(cth, "Myotube", classify_func = function(x) { x[MYH3_id,] >= 1 })
- cth <- addCellType(cth, "Reserve cell", classify_func = function(x) { x[MYH3_id,] == 0 & x[CCNB2_id,] == 0 })
- HSMM_myo <- classifyCells(HSMM_myo, cth)
- #寻找不同的细胞类型中与marker基因共同差异表达的top1000基因
- marker_diff <- markerDiffTable(HSMM_myo[HSMM_expressed_genes,], cth, cores = 1)
- semisup_clustering_genes <- row.names(marker_diff)[order(marker_diff$qval)][1:1000]
- #构建单细胞路径
- HSMM_myo <- setOrderingFilter(HSMM_myo, semisup_clustering_genes)
- #plot_ordering_genes(HSMM_myo)
- HSMM_myo <- reduceDimension(HSMM_myo, max_components = 2, method = 'DDRTree', norm_method = 'log')
- HSMM_myo <- orderCells(HSMM_myo)
- HSMM_myo <- orderCells(HSMM_myo, root_state = GM_state(HSMM_myo))
- plot_cell_trajectory(HSMM_myo, color_by = "CellType")
- theme(legend.position = "right")
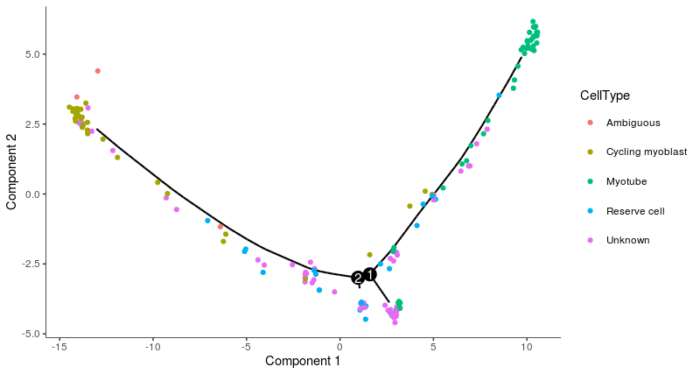
可以看到这张图中有两个分叉点,第二个分叉点较小,我们认为其为噪声,并不是具有真正的生物学意义。在分支1处,一部分细胞成功发育成成熟的肌管细胞,一部分细胞则未能成功实现完全分化,我们可以用特定的基因表达加以验证。
- #进一步验证伪时序分析
- HSMM_filtered <- HSMM_myo[HSMM_expressed_genes,]
- my_genes <- row.names(subset(fData(HSMM_filtered), gene_short_name %in% c("CDK1", "MEF2C", "MYH3")))
- cds_subset <- HSMM_filtered[my_genes,]
- plot_genes_branched_pseudotime(cds_subset, branch_point = 1, color_by = "Hours", ncol = 1)
差异表达分析
这教程也实在是太长了,搞了我好几天,终于进入了最后的差异表达分析环节,难受。差异表达分析基本是RNA-seq的必备环节了,由于单细胞RNA-seq数据量大,因此使用单CPU将耗费大量时间,为了保证本教程的快速高效,我们先选取几个基因的downsample set,来演示一下最基本的差异表达分析,再进一步介绍更多的功能。在基本的分析流程中,我们主要关注培养基变化前后,基因的差异表达,另外,我们所选择的downsample set的基因都是与肌肉发育高度相关的,因此显著性都会比较高。
- #downsample基因
- marker_genes <- row.names(subset(fData(HSMM_myo),
- gene_short_name %in% c("MEF2C", "MEF2D", "MYF5",
- "ANPEP", "PDGFRA","MYOG",
- "TPM1", "TPM2", "MYH2",
- "MYH3", "NCAM1", "TNNT1",
- "TNNT2", "TNNC1", "CDK1",
- "CDK2", "CCNB1", "CCNB2",
- "CCND1", "CCNA1", "ID1")))
- #按照培养基的不同进行差异表达分析
- diff_test_res <- differentialGeneTest(HSMM_myo[marker_genes,], fullModelFormulaStr = "~Media")
- #以FDR=10%为阈值进行筛选
- sig_genes <- subset(diff_test_res, qval < 0.1)
- sig_genes[,c("gene_short_name", "pval", "qval")]
由于这些基因都与肌肉发育相关,因此大部分都表现出了显著的差异表达。
- > sig_genes[,c("gene_short_name", "pval", "qval")]
- gene_short_name pval qval
- ENSG00000081189.9 MEF2C 7.216743e-23 3.788790e-22
- ENSG00000105048.12 TNNT1 9.651055e-13 2.533402e-12
- ENSG00000109063.9 MYH3 4.258280e-35 4.471194e-34
- ENSG00000111049.3 MYF5 1.410577e-34 9.874040e-34
- ENSG00000114854.3 TNNC1 3.489570e-18 1.221350e-17
- ENSG00000118194.14 TNNT2 1.499063e-42 3.148033e-41
- ENSG00000122180.4 MYOG 1.402932e-13 4.208796e-13
- ENSG00000123374.6 CDK2 9.465533e-03 1.242351e-02
- ENSG00000125414.13 MYH2 1.616586e-06 2.611409e-06
- ENSG00000125968.7 ID1 1.331859e-05 1.997788e-05
- ENSG00000134057.10 CCNB1 6.125710e-11 1.429332e-10
- ENSG00000134853.7 PDGFRA 3.288918e-02 4.062781e-02
- ENSG00000140416.15 TPM1 2.414145e-08 4.608823e-08
- ENSG00000149294.12 NCAM1 2.084944e-20 8.756763e-20
- ENSG00000157456.3 CCNB2 8.009724e-08 1.401702e-07
- ENSG00000166825.9 ANPEP 3.962394e-02 4.622794e-02
- ENSG00000170312.11 CDK1 2.139016e-08 4.491933e-08
- ENSG00000198467.8 TPM2 1.012980e-03 1.418173e-03
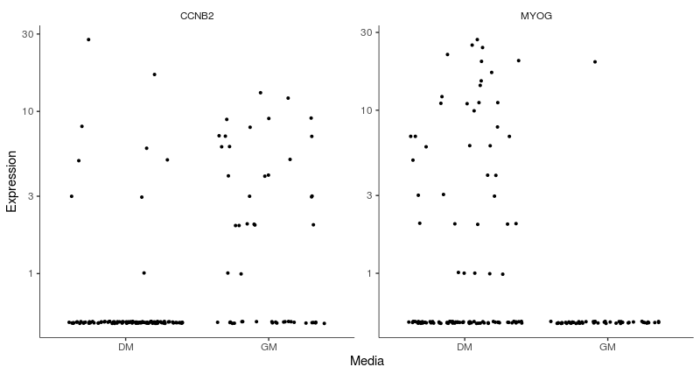
具体展示MYOG和CCNB2两个基因在培养基变化前后的表达情况。
- #具体展示MYOG和CCNB2两个基因在培养基变化前后的表达情况
- MYOG_ID1 <- HSMM_myo[row.names(subset(fData(HSMM_myo), gene_short_name %in% c("MYOG", "CCNB2"))),]
- plot_genes_jitter(MYOG_ID1, grouping = "Media", ncol= 2)
下面就从四个方面进一步展示Monocle2所提供的一系列差异表达分析的工具与方法。
寻找用于区分细胞类型或细胞状态的基因
在前面的分析中,我们仅仅使用了单一的marker基因来区分细胞类型,那我们是不是还能找到一些基因,来测试使用它们区分细胞类型的能力呢?这就是我们要在这一小节所做的分析。我们挑选三个基因UBC, NCAM1, ANPEP来做,我们先来看下这三个基因在两种细胞类型中的表达情况。
- #寻找用于区分细胞类型和细胞状态的基因
- to_be_tested <- row.names(subset(fData(HSMM), gene_short_name %in% c("UBC", "NCAM1", "ANPEP")))
- cds_subset <- HSMM[to_be_tested,]
- plot_genes_jitter(cds_subset, grouping = "CellType", color_by = "CellType", nrow= 1, ncol = NULL, plot_trend = TRUE)
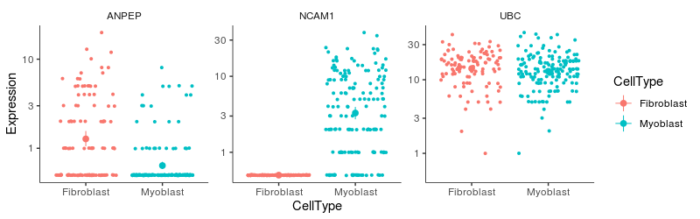
其中,管家基因UBC作为阴性对照,在两种细胞类型间的表达量差异不大,我们接下来的问题就是使用这三个基因中的一个基因来区分两种细胞类型的能力如何,这也就反映了细胞类型对该基因的表达的影响程度。Monocle2的想法是建立两个模型,一个叫full模型,提供我们想要区分的细胞类型的信息;还有一个叫reduced模型,不提供细胞类型的信息。这两个模型都能去预测某个基因在细胞中的表达量,但由于full模型提供了更多的信息,因此full模型的预测结果会比reduced模型要好,那么full模型比reduced模型预测准确率更好的程度(显著性)就反映了细胞类型的信息对基因表达的影响程度,也就反映了基因用于区分两种细胞的能力。
- #构建full和reduced模型进行比较
- diff_test_res <- differentialGeneTest(cds_subset, fullModelFormulaStr = "~CellType")
- diff_test_res[,c("gene_short_name", "pval", "qval")]
其中differentialGeneTest函数中reducedModelFormulaStr参数默认为‘~1’,不需要进行改动。
- > diff_test_res[,c("gene_short_name", "pval", "qval")]
- gene_short_name pval qval
- ENSG00000149294.12 NCAM1 4.505519e-92 1.351656e-91
- ENSG00000150991.10 UBC 4.000686e-01 4.000686e-01
- ENSG00000166825.9 ANPEP 4.007268e-13 6.010901e-13
可以看到NCAM1的显著性最高,UBC的显著性最低,这与上面的基因表达散点图是相吻合的。另外,还可以用另一组代码来实现,也是一样的。
- > #另一组代码实现
- > full_model_fits <- fitModel(cds_subset, modelFormulaStr = "~CellType")
- > reduced_model_fits <- fitModel(cds_subset, modelFormulaStr = "~1")
- > diff_test_res <- compareModels(full_model_fits, reduced_model_fits)
- > diff_test_res
- status family pval
- ENSG00000149294.12 OK negbinomial.size 4.505519e-92
- ENSG00000150991.10 OK negbinomial.size 4.000686e-01
- ENSG00000166825.9 OK negbinomial.size 4.007268e-13
- qval
- ENSG00000149294.12 1.351656e-91
- ENSG00000150991.10 4.000686e-01
- ENSG00000166825.9 6.010901e-13
基因随伪时序变化的规律
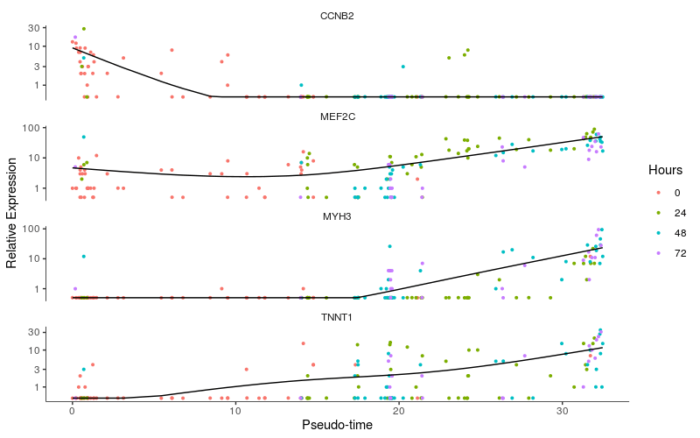
和上一节类似,上一节中我们构建的是基因随着细胞类型或状态发生变化的函数,在这一节中我们构建的是基因随伪时序变化的函数,因此在这一节的分析中,我们既可以探究伪时序变化对基因表达影响的显著性,已经基因随伪时序的变化规律,我们用MYH3, MEF2C, CCNB2, TNNT1这四个基因加以说明。
- #基因随伪时序变化的规律
- to_be_tested <- row.names(subset(fData(HSMM), gene_short_name %in% c("MYH3", "MEF2C", "CCNB2", "TNNT1")))
- cds_subset <- HSMM_myo[to_be_tested,]
- diff_test_res <- differentialGeneTest(cds_subset, fullModelFormulaStr = "~sm.ns(Pseudotime)")
- diff_test_res[,c("gene_short_name", "pval", "qval")]
注意这里的模型选择,”~sm.ns(Pseudotime)”表示模型将拟合出一条平滑的曲线去表征基因表达随伪时序变化的函数。
- > diff_test_res[,c("gene_short_name", "pval", "qval")]
- gene_short_name pval qval
- ENSG00000081189.9 MEF2C 6.652142e-43 1.330428e-42
- ENSG00000105048.12 TNNT1 2.171522e-31 2.895363e-31
- ENSG00000109063.9 MYH3 2.365136e-73 9.460543e-73
- ENSG00000157456.3 CCNB2 1.267467e-27 1.267467e-27
我们进一步画出基因表达随伪时序变化的曲线,来验证上述结果。
- #基因表达随伪时序变化的曲线
- plot_genes_in_pseudotime(cds_subset, color_by = "Hours")
根据伪时序表达规律对基因进行聚类
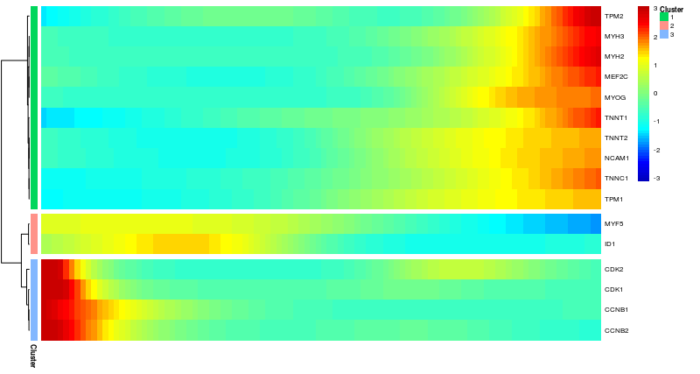
我们可能会问这样一个问题:哪些基因在生物学过程中有相似的动力学特征?Monocle2提供了根据伪时序表达规律对基因进行聚类的功能,让我们能将这些基因进行分组去观察他们的共性,并以热图的形式展示出来。
- #以随伪时序变化的规律对基因进行聚类
- diff_test_res <- differentialGeneTest(HSMM_myo[marker_genes,], fullModelFormulaStr = "~sm.ns(Pseudotime)")
- sig_gene_names <- row.names(subset(diff_test_res, qval < 0.1))
- plot_pseudotime_heatmap(HSMM_myo[sig_gene_names,], num_clusters = 3, cores = 1, show_rownames = T)
多因素差异表达分析
Monocle2允许在多个因子存在的情况下进行差异表达分析,从而减去一些因子的效果观察剩下的因子对基因表达的影响(感觉和ANOVA差不多)。在下面的例子中,选择full模型考虑细胞类型和时间,reduced模型考虑时间,从而将时间的影响排除掉,仅仅考察细胞类型对基因表达的影响。
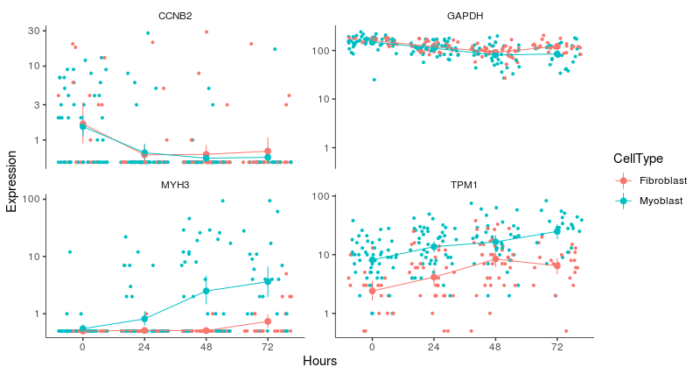
- #多因素差异表达分析
- to_be_tested <- row.names(subset(fData(HSMM), gene_short_name %in% c("TPM1", "MYH3", "CCNB2", "GAPDH")))
- cds_subset <- HSMM[to_be_tested,]
- diff_test_res <- differentialGeneTest(cds_subset, fullModelFormulaStr = "~CellType Hours", reducedModelFormulaStr = "~Hours")
- diff_test_res[,c("gene_short_name", "pval", "qval")]
- > diff_test_res[,c("gene_short_name", "pval", "qval")]
- gene_short_name pval qval
- ENSG00000109063.9 MYH3 4.990678e-55 1.996271e-54
- ENSG00000111640.10 GAPDH 1.974650e-01 1.974650e-01
- ENSG00000140416.15 TPM1 2.540899e-22 5.081798e-22
- ENSG00000157456.3 CCNB2 1.358945e-02 1.811926e-02
再结合散点图来看下表达情况加以验证。
- #基因表达散点图
- plot_genes_jitter(cds_subset,grouping = "Hours", color_by = "CellType", plot_trend = TRUE)
- facet_wrap( ~ feature_label, scales= "free_y")
分析单细胞路径的分支
通常,Monocle2所构建的单细胞路径会引入分支,因为细胞执行了不同的基因表达程序,分支点因此也就成了细胞命运的转折点值得我们仔细分析,Monocle2提供了一系列方法便利我们的分析。这里的分析使用的是一组肺的数据,数据捕捉到了早期发育过程中的细胞,晚期的AT1和AT2型细胞,以及一些中间态的细胞。
- #单细胞路径的分支分析
- #导入数据
- lung <- load_lung()
- plot_cell_trajectory(lung, color_by = "Time")
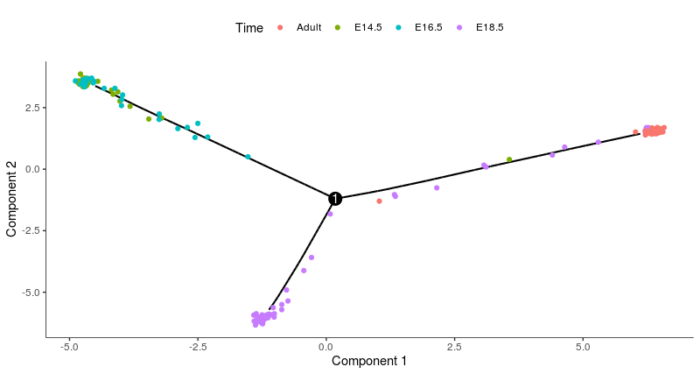
上图展示了从这些数据构建的单细胞路径,包含一个分支。自然而然地我们也会提出这样的问题,从发育早期到分支点处,哪些基因发生了变化?两个分支的细胞基因表达上有什么差异?我们单细胞路径分支的分析便是要解决这些问题。
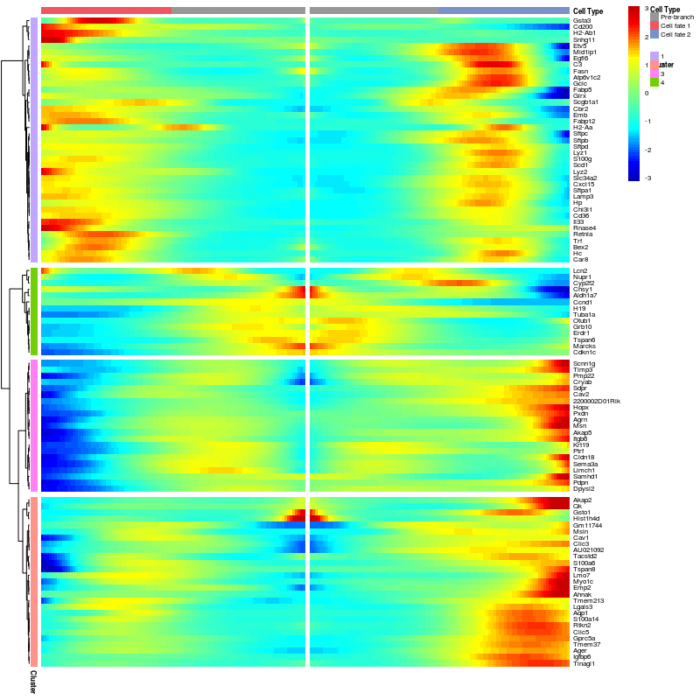
BEAM函数吃入一个已经构建好单细胞路径的CellDataSet对象和分支点名称,返回对各个基因的显著性评估,基因的显著性越高,则分支的形成就越依赖于他们的表达。
- #基因表达对分支形成的显著性
- BEAM_res <- BEAM(lung, branch_point = 1, cores = 1)
- BEAM_res <- BEAM_res[order(BEAM_res$qval),]
- BEAM_res <- BEAM_res[,c("gene_short_name", "pval", "qval")]
用热图的形式展示两个lineage的基因表达变化与基因表达变化的聚类分析。
- #用热图的形式展示两个lineage的基因表达变化与基因表达变化的聚类分析
- plot_genes_branched_heatmap(lung[row.names(subset(BEAM_res, qval < 1e-4)),], branch_point = 1, num_clusters = 4, cores = 1, use_gene_short_name = T, show_rownames = T)
我们可以进一步挑一些体系中比较重要的基因,沿着伪时序,按照两个lineage,展示其基因表达的变化趋势。
- #重要基因沿伪时序的表达变化,分支对比
- lung_genes <- row.names(subset(fData(lung), gene_short_name %in% c("Ccnd2", "Sftpb", "Pdpn")))
- plot_genes_branched_pseudotime(lung[lung_genes,], branch_point = 1, color_by = "Time", ncol = 1)
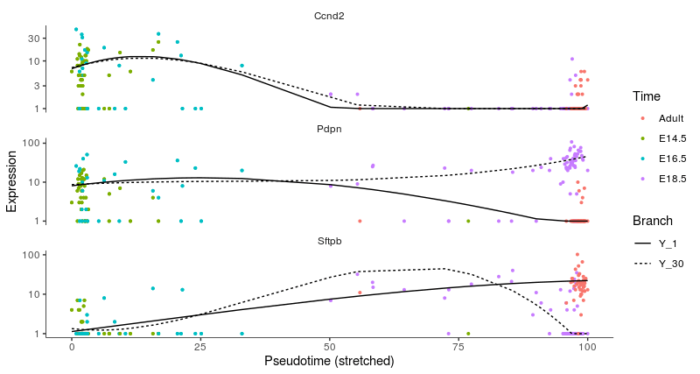
可以看到CCND2作为一种细胞周期基因,在两个lineage上均显示出了持续下降的趋势,并且重合程度高,因此在BEAM测试中并不显著。最后老规矩,贴一个完整代码。
- setwd('/your/path/to/monocle')
- #导入包
- library(monocle)
- library(HSMMSingleCell)
- library(reshape2)
- data("HSMM_expr_matrix")
- data("HSMM_gene_annotation")
- data("HSMM_sample_sheet")
- #使用相对表达举证先创建一个CellDataSet对象
- pd <- new("AnnotatedDataFrame", data = HSMM_sample_sheet)
- fd <- new("AnnotatedDataFrame", data = HSMM_gene_annotation)
- HSMM <- newCellDataSet(as.matrix(HSMM_expr_matrix),phenoData = pd,featureData = fd,lowerDetectionLimit = 0.1,expressionFamily = tobit(Lower = 0.1))
- #使用CellDataSet估计RNA counts数
- rpc_matrix <- relative2abs(HSMM, method = "num_genes")
- #使用RNA counts数构建一个新的CellDataSet对象
- HSMM <- newCellDataSet(as(as.matrix(rpc_matrix), "sparseMatrix"),phenoData = pd,featureData = fd,lowerDetectionLimit = 0.5,expressionFamily = negbinomial.size())
- #估计数据集大小因子和离散程度
- HSMM <- estimateSizeFactors(HSMM)
- HSMM <- estimateDispersions(HSMM)
- #查看每个基因都有多少细胞表达
- HSMM <- detectGenes(HSMM, min_expr = 0.1)
- print(head(fData(HSMM)))
- #以10个细胞为阈值进行筛选
- expressed_genes <- row.names(subset(fData(HSMM),num_cells_expressed >= 10))
- #对细胞进行筛选
- print(head(pData(HSMM)))
- #统计各个细胞中mRNA的总表达水平
- pData(HSMM)$Total_mRNAs <- Matrix::colSums(exprs(HSMM))
- HSMM <- HSMM[,pData(HSMM)$Total_mRNAs < 1e6]
- qplot(Total_mRNAs, data = pData(HSMM), color = Hours, geom ="density")
- #以两倍标准差作为上下限的阈值
- upper_bound <- 10^(mean(log10(pData(HSMM)$Total_mRNAs)) 2*sd(log10(pData(HSMM)$Total_mRNAs)))
- lower_bound <- 10^(mean(log10(pData(HSMM)$Total_mRNAs)) - 2*sd(log10(pData(HSMM)$Total_mRNAs)))
- qplot(Total_mRNAs, data = pData(HSMM), color = Hours, geom ="density")
- geom_vline(xintercept = lower_bound)
- geom_vline(xintercept = upper_bound)
- #筛选,并注意由于剔除了部分细胞,因此基因的相关数据要重新计算
- HSMM <- HSMM[,pData(HSMM)$Total_mRNAs > lower_bound & pData(HSMM)$Total_mRNAs < upper_bound]
- HSMM <- detectGenes(HSMM, min_expr = 0.1)
- #验证筛选过后,细胞的mRNA表达水平是否为对数正态分布
- #对mRNA表达水平取log
- L <- log(exprs(HSMM[expressed_genes,]))
- #对每个基因进行标准化,并使用melt函数改变数据形式
- melted_dens_df <- melt(Matrix::t(scale(Matrix::t(L))))
- #作图
- qplot(value, geom = "density", data = melted_dens_df)
- stat_function(fun = dnorm, size = 0.5, color = 'red')
- xlab("Standardized log(FPKM)")
- ylab("Density")
- #鉴定细胞类型
- MYF5_id <- row.names(subset(fData(HSMM), gene_short_name == "MYF5"))
- ANPEP_id <- row.names(subset(fData(HSMM),gene_short_name == "ANPEP"))
- #构建cth类,确定筛选规则
- cth <- newCellTypeHierarchy()
- cth <- addCellType(cth, "Myoblast", classify_func = function(x) { x[MYF5_id,] >= 1 })
- cth <- addCellType(cth, "Fibroblast", classify_func = function(x) { x[MYF5_id,] < 1 & x[ANPEP_id,] > 1 })
- #通过classifyCells函数给phenoData加入新的一列:CellType
- HSMM <- classifyCells(HSMM, cth, 0.1)
- #查看筛选情况
- table(pData(HSMM)$CellType)
- #pie图可视化
- pie <- ggplot(pData(HSMM), aes(x = factor(1), fill = factor(CellType))) geom_bar(width = 1)
- pie coord_polar(theta = "y") theme(axis.title.x = element_blank(), axis.title.y = element_blank())
- #无监督的方法对细胞进行聚类
- #筛选出有一定表达量的基因
- disp_table <- dispersionTable(HSMM)
- unsup_clustering_genes <- subset(disp_table, mean_expression >= 0.1)
- HSMM <- setOrderingFilter(HSMM, unsup_clustering_genes$gene_id)
- plot_ordering_genes(HSMM)
- #t-SNE降维
- #线性降维选择PCA,分析不同的PC轴对变异的贡献,默认选择log函数进行标准化
- plot_pc_variance_explained(HSMM, return_all = F)
- #选择前6个PC进行降维聚类
- HSMM <- reduceDimension(HSMM, max_components = 2, num_dim = 6, reduction_method = 'tSNE', verbose = T)
- HSMM <- clusterCells(HSMM, num_clusters = 2)
- plot_cell_clusters(HSMM, 1, 2, color = "CellType")
- #培养基引入的差异
- plot_cell_clusters(HSMM, 1, 2, color = "Media")
- #减去培养基和基因表达量所引入的差异
- HSMM <- reduceDimension(HSMM, max_components = 2, num_dim = 2, reduction_method = 'tSNE', residualModelFormulaStr = "~Media num_genes_expressed", verbose = T)
- HSMM <- clusterCells(HSMM, num_clusters= 2 )
- plot_cell_clusters(HSMM, 1, 2, color = "CellType")
- #从三种细胞类型来看聚类情况
- HSMM <- clusterCells(HSMM, num_clusters = 2 )
- plot_cell_clusters(HSMM, 1, 2, color = "Cluster") facet_wrap(~CellType)
- #半监督的方法对基因进行聚类
- #去除培养基和基因数的差异,计算出和marker基因共同差异表达的基因
- marker_diff <- markerDiffTable(HSMM[expressed_genes,], cth, residualModelFormulaStr = "~Media num_genes_expressed", cores = 1)
- #对显著性和细胞类型的特异性进行筛选
- candidate_clustering_genes <- row.names(subset(marker_diff, qval < 0.01))
- marker_spec <- calculateMarkerSpecificity(HSMM[candidate_clustering_genes,], cth)
- head(selectTopMarkers(marker_spec, 3))
- #为每种细胞类型挑选最特异的500个基因用于后续的降维聚类
- semisup_clustering_genes <- unique(selectTopMarkers(marker_spec, 500)$gene_id)
- HSMM <- setOrderingFilter(HSMM, semisup_clustering_genes)
- plot_ordering_genes(HSMM)
- #t-SNE降维并聚类
- #PC轴对差异的贡献
- plot_pc_variance_explained(HSMM, return_all = F)
- #t-SNE
- HSMM <- reduceDimension(HSMM, max_components = 2, num_dim = 3, norm_method = 'log', reduction_method = 'tSNE', residualModelFormulaStr = "~Media num_genes_expressed", verbose = T)
- HSMM <- clusterCells(HSMM, num_clusters = 2)
- plot_cell_clusters(HSMM, 1, 2, color = "CellType")
- #从三种细胞类型来看聚类情况
- HSMM <- clusterCells(HSMM, num_clusters = 2)
- plot_cell_clusters(HSMM, 1, 2, color = "Cluster") facet_wrap(~CellType)
- #将Unknown的细胞进行划分
- HSMM <- clusterCells(HSMM, num_clusters = 2, frequency_thresh = 0.1, cell_type_hierarchy = cth)
- plot_cell_clusters(HSMM, 1, 2, color = "CellType")
- #看下两种细胞的比例
- pie <- ggplot(pData(HSMM), aes(x = factor(1), fill = factor(CellType)))
- geom_bar(width = 1)
- pie
- coord_polar(theta = "y")
- theme(axis.title.x = element_blank(), axis.title.y = element_blank())
- #仅取成肌细胞用作后续的分析
- HSMM_myo <- HSMM[,pData(HSMM)$CellType == 'Myoblast']
- #构建单细胞路径
- #挑选用于后续构建路径的基因
- #无监督的算法,首尾两种细胞的差异表达基因
- diff_test_res <- differentialGeneTest(HSMM_myo[expressed_genes,], fullModelFormulaStr = "~Media")
- ordering_genes <- row.names (subset(diff_test_res, qval < 0.01))
- HSMM_myo <- setOrderingFilter(HSMM_myo, ordering_genes)
- plot_ordering_genes(HSMM_myo)
- #降维
- HSMM_myo <- reduceDimension(HSMM_myo, max_components = 2, method = 'DDRTree')
- #构建路径并可视化
- HSMM_myo <- orderCells(HSMM_myo)
- plot_cell_trajectory(HSMM_myo, color_by = "Hours")
- #树形结构state
- plot_cell_trajectory(HSMM_myo, color_by = "State")
- #判断那种state含有更多的0小时的细胞从而确定根
- GM_state <- function(cds){
- if (length(unique(pData(cds)$State)) > 1){
- T0_counts <- table(pData(cds)$State, pData(cds)$Hours)[,"0"]
- return(as.numeric(names(T0_counts)[which(T0_counts == max(T0_counts))]))
- } else {
- return (1)
- }
- }
- HSMM_myo <- orderCells(HSMM_myo, root_state = GM_state(HSMM_myo))
- plot_cell_trajectory(HSMM_myo, color_by = "Pseudotime")
- #分页显示不同的state
- plot_cell_trajectory(HSMM_myo, color_by = "State")
- facet_wrap(~State, nrow = 1)
- #从marker基因的表达来确定哪个state为根
- blast_genes <- row.names(subset(fData(HSMM_myo), gene_short_name %in% c("CCNB2", "MYOD1", "MYOG")))
- plot_genes_jitter(HSMM_myo[blast_genes,], grouping = "State", min_expr = 0.1)
- #marker基因随伪时序的变化规律,进一步验证可靠性
- HSMM_expressed_genes <- row.names(subset(fData(HSMM_myo), num_cells_expressed >= 10))
- HSMM_filtered <- HSMM_myo[HSMM_expressed_genes,]
- my_genes <- row.names(subset(fData(HSMM_filtered), gene_short_name %in% c("CDK1", "MEF2C", "MYH3")))
- cds_subset <- HSMM_filtered[my_genes,]
- plot_genes_in_pseudotime(cds_subset, color_by = "Hours")
- #其他的挑选用于构建单细胞路径的基因的方法
- #dqFeature法
- #t-SNE降维
- #先挑选出至少在5%的细胞中有表达的基因
- HSMM_myo <- detectGenes(HSMM_myo, min_expr = 0.1)
- fData(HSMM_myo)$use_for_ordering <- fData(HSMM_myo)$num_cells_expressed > 0.05 * ncol(HSMM_myo)
- #PCA主成分分析
- plot_pc_variance_explained(HSMM_myo, return_all = F)
- #选择前4个PC用于t-SNE降维
- HSMM_myo <- reduceDimension(HSMM_myo, max_components = 2, norm_method = 'log', num_dim = 4, reduction_method = 'tSNE', verbose = T)
- #默认参数聚类
- HSMM_myo <- clusterCells(HSMM_myo, verbose = F)
- #可视化
- plot_cell_clusters(HSMM_myo, color_by = 'as.factor(Cluster)')
- plot_cell_clusters(HSMM_myo, color_by = 'as.factor(Hours)')
- #调整参数
- plot_rho_delta(HSMM_myo)
- #选择delta阈值为4,rho阈值为3
- plot_rho_delta(HSMM_myo, rho_threshold = 3, delta_threshold = 4)
- #聚类,可视化
- HSMM_myo <- clusterCells(HSMM_myo, rho_threshold = 3, delta_threshold = 4, skip_rho_sigma = T, verbose = F)
- plot_cell_clusters(HSMM_myo, color_by = 'as.factor(Cluster)')
- plot_cell_clusters(HSMM_myo, color_by = 'as.factor(Hours)')
- #选择delta阈值为5,rho阈值为2
- HSMM_myo <- clusterCells(HSMM_myo, rho_threshold = 2, delta_threshold = 5, skip_rho_sigma = T, verbose = F)
- plot_cell_clusters(HSMM_myo, color_by = 'as.factor(Cluster)')
- plot_cell_clusters(HSMM_myo, color_by = 'as.factor(Hours)')
- #计算cluster之间的差异表达基因
- clustering_DEG_genes <- differentialGeneTest(HSMM_myo[HSMM_expressed_genes,], fullModelFormulaStr = '~Cluster', cores = 1)
- #选择前1000个最显著的差异表达基因用于构建单细胞路径
- HSMM_ordering_genes <- row.names(clustering_DEG_genes)[order(clustering_DEG_genes$qval)][1:1000]
- HSMM_myo <- setOrderingFilter(HSMM_myo, ordering_genes = HSMM_ordering_genes)
- HSMM_myo <- reduceDimension(HSMM_myo, method = 'DDRTree')
- HSMM_myo <- orderCells(HSMM_myo)
- HSMM_myo <- orderCells(HSMM_myo, root_state = GM_state(HSMM_myo))
- plot_cell_trajectory(HSMM_myo, color_by = "Hours")
- #半监督算法,CCNB2作为早期成肌细胞的marker,MYH3作为晚期肌管细胞的marker
- CCNB2_id <- row.names(subset(fData(HSMM_myo), gene_short_name == "CCNB2"))
- MYH3_id <- row.names(subset(fData(HSMM_myo), gene_short_name == "MYH3"))
- cth <- newCellTypeHierarchy()
- cth <- addCellType(cth, "Cycling myoblast", classify_func = function(x) { x[CCNB2_id,] >= 1 })
- cth <- addCellType(cth, "Myotube", classify_func = function(x) { x[MYH3_id,] >= 1 })
- cth <- addCellType(cth, "Reserve cell", classify_func = function(x) { x[MYH3_id,] == 0 & x[CCNB2_id,] == 0 })
- HSMM_myo <- classifyCells(HSMM_myo, cth)
- #寻找不同的细胞类型中与marker基因共同差异表达的top1000基因
- marker_diff <- markerDiffTable(HSMM_myo[HSMM_expressed_genes,], cth, cores = 1)
- semisup_clustering_genes <- row.names(marker_diff)[order(marker_diff$qval)][1:1000]
- #构建单细胞路径
- HSMM_myo <- setOrderingFilter(HSMM_myo, semisup_clustering_genes)
- HSMM_myo <- reduceDimension(HSMM_myo, max_components = 2, method = 'DDRTree', norm_method = 'log')
- HSMM_myo <- orderCells(HSMM_myo)
- HSMM_myo <- orderCells(HSMM_myo, root_state = GM_state(HSMM_myo))
- plot_cell_trajectory(HSMM_myo, color_by = "CellType")
- theme(legend.position = "right")
- #进一步验证伪时序分析
- HSMM_filtered <- HSMM_myo[HSMM_expressed_genes,]
- my_genes <- row.names(subset(fData(HSMM_filtered), gene_short_name %in% c("CDK1", "MEF2C", "MYH3")))
- cds_subset <- HSMM_filtered[my_genes,]
- plot_genes_branched_pseudotime(cds_subset, branch_point = 1, color_by = "Hours", ncol = 1)
- #差异表达分析
- #downsample基因
- marker_genes <- row.names(subset(fData(HSMM_myo),
- gene_short_name %in% c("MEF2C", "MEF2D", "MYF5",
- "ANPEP", "PDGFRA","MYOG",
- "TPM1", "TPM2", "MYH2",
- "MYH3", "NCAM1", "TNNT1",
- "TNNT2", "TNNC1", "CDK1",
- "CDK2", "CCNB1", "CCNB2",
- "CCND1", "CCNA1", "ID1")))
- #按照培养基的不同进行差异表达分析
- diff_test_res <- differentialGeneTest(HSMM_myo[marker_genes,], fullModelFormulaStr = "~Media")
- #以FDR=10%为阈值进行筛选
- sig_genes <- subset(diff_test_res, qval < 0.1)
- sig_genes[,c("gene_short_name", "pval", "qval")]
- #具体展示MYOG和CCNB2两个基因在培养基变化前后的表达情况
- MYOG_ID1 <- HSMM_myo[row.names(subset(fData(HSMM_myo), gene_short_name %in% c("MYOG", "CCNB2"))),]
- plot_genes_jitter(MYOG_ID1, grouping = "Media", ncol= 2)
- #寻找用于区分细胞类型和细胞状态的基因
- to_be_tested <- row.names(subset(fData(HSMM), gene_short_name %in% c("UBC", "NCAM1", "ANPEP")))
- cds_subset <- HSMM[to_be_tested,]
- plot_genes_jitter(cds_subset, grouping = "CellType", color_by = "CellType", nrow= 1, ncol = NULL, plot_trend = TRUE)
- #构建full和reduced模型进行比较
- diff_test_res <- differentialGeneTest(cds_subset, fullModelFormulaStr = "~CellType")
- diff_test_res[,c("gene_short_name", "pval", "qval")]
- #另一组代码实现
- full_model_fits <- fitModel(cds_subset, modelFormulaStr = "~CellType")
- reduced_model_fits <- fitModel(cds_subset, modelFormulaStr = "~1")
- diff_test_res <- compareModels(full_model_fits, reduced_model_fits)
- diff_test_res
- #基因随伪时序变化的规律
- to_be_tested <- row.names(subset(fData(HSMM), gene_short_name %in% c("MYH3", "MEF2C", "CCNB2", "TNNT1")))
- cds_subset <- HSMM_myo[to_be_tested,]
- diff_test_res <- differentialGeneTest(cds_subset, fullModelFormulaStr = "~sm.ns(Pseudotime)")
- diff_test_res[,c("gene_short_name", "pval", "qval")]
- #基因表达随伪时序变化的曲线
- plot_genes_in_pseudotime(cds_subset, color_by = "Hours")
- #以随伪时序变化的规律对基因进行聚类
- diff_test_res <- differentialGeneTest(HSMM_myo[marker_genes,], fullModelFormulaStr = "~sm.ns(Pseudotime)")
- sig_gene_names <- row.names(subset(diff_test_res, qval < 0.1))
- plot_pseudotime_heatmap(HSMM_myo[sig_gene_names,], num_clusters = 3, cores = 1, show_rownames = T)
- #多因素差异表达分析
- to_be_tested <- row.names(subset(fData(HSMM), gene_short_name %in% c("TPM1", "MYH3", "CCNB2", "GAPDH")))
- cds_subset <- HSMM[to_be_tested,]
- diff_test_res <- differentialGeneTest(cds_subset, fullModelFormulaStr = "~CellType Hours", reducedModelFormulaStr = "~Hours")
- diff_test_res[,c("gene_short_name", "pval", "qval")]
- #基因表达散点图
- plot_genes_jitter(cds_subset,grouping = "Hours", color_by = "CellType", plot_trend = TRUE)
- facet_wrap( ~ feature_label, scales= "free_y")
- #单细胞路径的分支分析
- #导入数据
- lung <- load_lung()
- plot_cell_trajectory(lung, color_by = "Time")
- #基因表达对分支形成的显著性
- BEAM_res <- BEAM(lung, branch_point = 1, cores = 1)
- BEAM_res <- BEAM_res[order(BEAM_res$qval),]
- BEAM_res <- BEAM_res[,c("gene_short_name", "pval", "qval")]
- #用热图的形式展示两个lineage的基因表达变化与基因表达变化的聚类分析
- plot_genes_branched_heatmap(lung[row.names(subset(BEAM_res, qval < 1e-4)),], branch_point = 1, num_clusters = 4, cores = 1, use_gene_short_name = T, show_rownames = T)
- #重要基因沿伪时序的表达变化,分支对比
- lung_genes <- row.names(subset(fData(lung), gene_short_name %in% c("Ccnd2", "Sftpb", "Pdpn")))
- plot_genes_branched_pseudotime(lung[lung_genes,], branch_point = 1, color_by = "Time", ncol = 1)