这一篇文章中主要是使用David进行GO分析的数据处理流程和简单的代码,具体GO分析的原理就不再涉及。
导出基因列表
- #基因列表的导出
- gene_cluster <- rownames(as.matrix(temp_gene[temp_gene[,"Cluster"] == '1',]))
- GO <- file('GO.txt','w')
- for (i in 1:length(gene_cluster)){
- writeLines(as.character(gene_cluster[i]),GO)
- }
- close(GO)
得到下面的一个txt文件,包含了我们感兴趣的基因。
David进行GO分析
进入David首页,选择functional annotation,提交基因list。
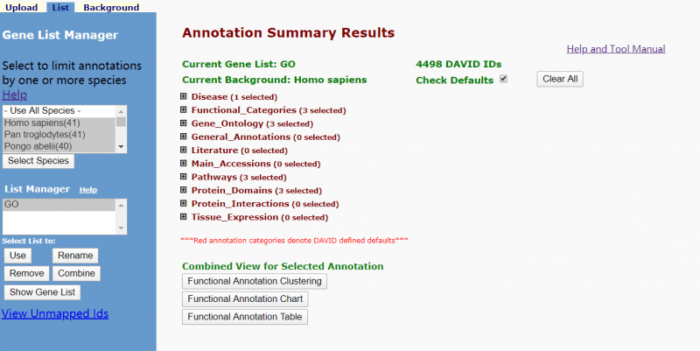
David会返回一个gene list的读取报告,比如gene symbol在各个物种中的map情况等,注意current background选择对应的物种。
进入functional annotation chart,对基因的功能在各种通路中进行富集,如GO的三大块(BP,CC,MF)以及KEGG等。
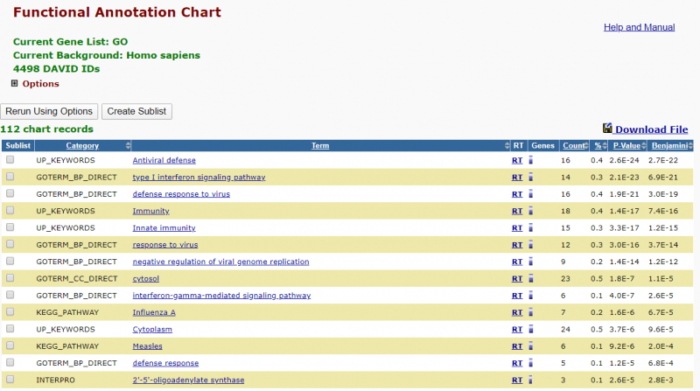
可以看到返回了各种富集的Term,以及显著性水平等信息,我一般会在option里把fold enrichment和FDR也选上,作为对显著性的评估。点击download file,复制转跳出来的全部网页内容,黏贴到GO.txt中。

download file转跳出来大概就长这样,复制后黏贴到txt文本里,可以使用之前的GO.txt,把原来的gene list覆盖就行。新建一个excel,数据自文本导入GO.txt。
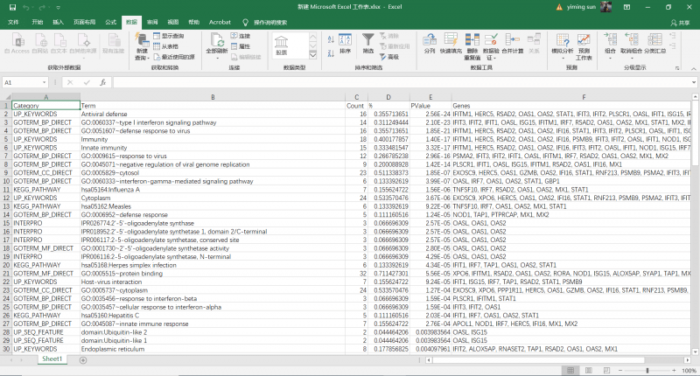
导出cvs格式即可,其实就是为了将字符转换为UTF-8,防止R读取后出现乱码而已。
R筛选相关的Term
R读取csv表格后,对数据框进行简单的筛选和排序。
- #简单的函数来执行
- GO_filter <- function(file,term = 'GOTERM_BP'){
- gene_cluster <- read.csv(file = file,header = T)
- gene_cluster <- gene_cluster[grep(pattern = term,gene_cluster$Category),]
- if(grepl(pattern = 'GO',term)){
- gene_cluster$Term <- sub(pattern = '^GO.*~','',gene_cluster$Term)
- }
- if(grepl(pattern = 'KEGG',term)){
- gene_cluster$Term <- sub(pattern = '^hsa.*:','',gene_cluster$Term)
- }
- gene_cluster <- gene_cluster[order(gene_cluster$FDR,decreasing = F),]
- return(gene_cluster)
- }
- #调用函数即可
- gene_cluster1 <- GO_filter(file = 'GO.csv')
后面只需要根据上面的操作,类似地构建gene_cluster2, gene_cluster3……即可。
ggplot2可视化
最后再使用ggplot2进行可视化即可,这里先提供两组代码,一组是多个cluster的分页柱状图显示,另一组是GO气泡图。后面会继续更新代码,如果我画出了更好看的GO图的话。
分页柱状图
分页柱状图,就是以分页的方式,把多组GO分析的统计图画在一起。
- #分页柱状图
- #先合并GO分析的表格,并在group列注释清楚
- gene_cluster <- rbind(gene_cluster1[1:5,],gene_cluster2[1:5,],gene_cluster3[1:5,])
- rownames(gene_cluster) <- 1:15
- gene_cluster <- cbind(gene_cluster,group = rep(c('gene_cluster1','gene_cluster2','gene_cluster3'),c(5,5,5)))
- #p_value取-log10
- gene_cluster[,"PValue"] <- -log10(gene_cluster[,'PValue'])
- #facet_wrap实现分页
- ggplot(data = gene_cluster,aes(x=Term,y=PValue))
- geom_bar(stat = 'identity',width = 0.8,fill = '#00b2ee')
- facet_wrap(~group,nrow = 3,scales = 'free_y')
- xlab('')
- ylab('-log10(pvalue)')
- coord_flip()
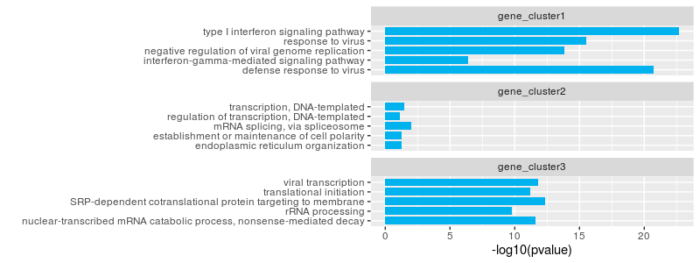
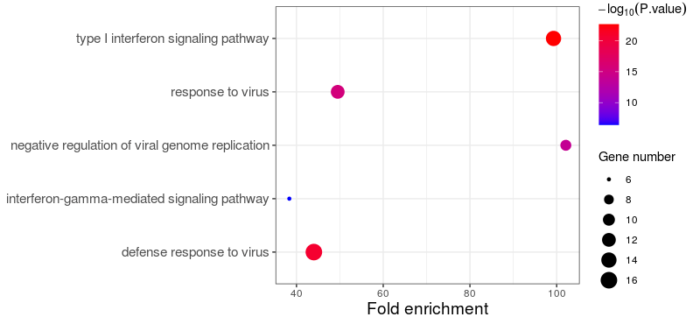
GO气泡图
- #GO气泡图
- #注意,这里gene_cluster1的PValue还未取-log10
- ggplot(gene_cluster1,aes(x=Fold.Enrichment,y=Term))
- geom_point(aes(size=Count,color=-1*log10(PValue)))
- scale_colour_gradient(low="blue",high="red")
- labs(
- color=expression(-log[10](P.value)),
- size="Gene number",
- x="Fold enrichment",
- y="Pathway name")
- theme_bw()
- theme(
- axis.text.y = element_text(size = rel(1.3)),
- axis.title.x = element_text(size=rel(1.3)),
- axis.title.y = element_blank()
- )