

DESeq2是比较常用的bulk RNA-seq分析软件,也可以处理类似的ChIP-Seq,shRNA以及质谱数据。DESeq2基于负二项分布的模型对count table进行数据处理,因此需要的输入是原始的counts,而不能预先进行标准化。本文着重于DESeq2分析前的准备,包括从SRA下载数据,使用STAR和Salmon进行mapping,构建DESeqDataSet等主要步骤。本文中用到的大部分包都维护在Bioconductor中,Bioconductor是专门用于处理基因组学数据的软件仓库,并协调不同软件之间的互相适配和调用(所以被称为conductor),例如下文中提到的SummarizedExperiment类就是多个Bioconductor包之间互相兼容的express table metadata通用对象。
简介
本文分析所用的实验数据是人类呼吸道平滑肌细胞,一组使用地塞米松进行处理,一组则不进行处理(地塞米松是一种具有抗炎症作用的合成糖皮质激素,常用于哮喘患者)。实验共使用了4种细胞系,因此共8个样本。实验数据可以从GSE52778中下载,也可以从Bioconductor的airway包中得到。本文将使用airway包简单讲解SummarizedExperiment类,再从GEO数据库中下载数据,分别使用STAR和Salmon进行mapping,再导入进DESeq2。STAR和Salmon是两种核酸测序读段比对软件,分别对应于两大类比对策略:比对到参考基因组以及比对到参考转录组。由于转录生成的RNA需要经过一系列的剪切修饰,因此一段RNA并不一定能完整地比对到基因组上,而是要将其拆分成不同的exon分别比对到基因组的相应位置上。因此比对到参考基因组的策略,包含了所有外显子的不同排列组合的可能,而比对到参考转录组的策略,则只能以所有已知的转录本作为参考。简而言之,比对参考基因组能够发现新的转录本,代价是计算开销大;比对到参考转录组则不能发现新的转录本,计算开销小。另外,通过直接比对到参考转录组,Salmon也可以得到测序转录本有效长度的信息,从而消除一部分bias。综上,目前bioconductor推荐使用Salmon tximport的方式对测序转录本进行定量并导入DESeq2,优点如下:
- 能够消除不同样本间基因长度不同的潜在差异。
- 计算开销相对较小。
- 避免丢弃过多可以比对到参考基因组上多个同源位点的转录本。
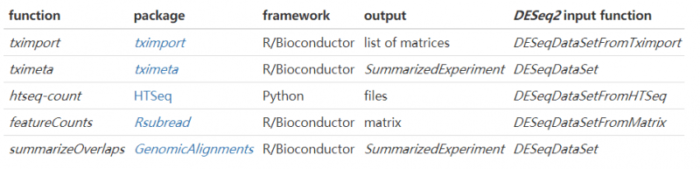
更多转录本比对策略的优缺点可以参考文献Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis。本文只介绍STAR summarizeOverlaps以及Salmon tximport将测序转录本导入DESeq2的方法,更多的方法见下图。
SummarizedExperiment类
上文提到SummarizedExperiment类是bioconductor中较为通用的express table metadata对象,这里我们简单探索一下这个数据结构,示意图如下。
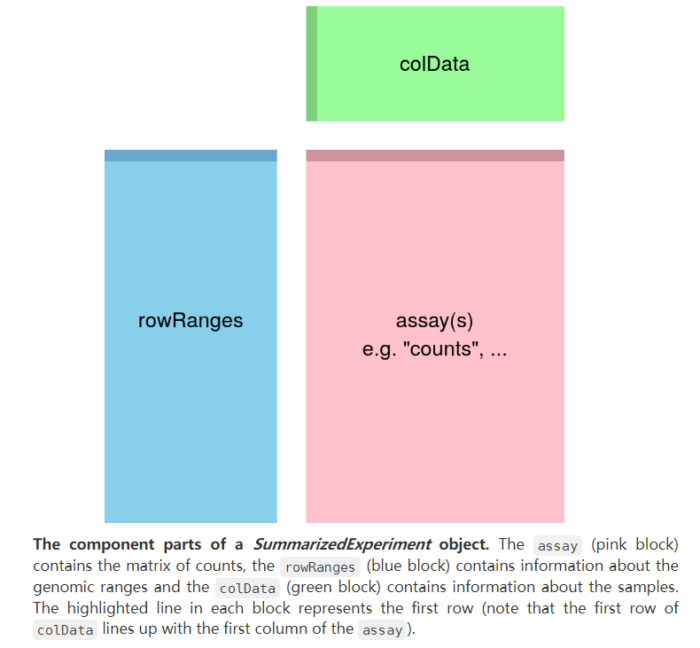
粉色部分assays包含了count matrix等信息,绿色部分colData包含了sample的metadata信息,蓝色部分rowRanges包含了feature的信息。assay中的matrix每一行为一个feature(gene),每一列为一个sample,sample和feature的metadata则对应于colData和rowRanges。下面我们使用airway包中整合好的数据来演示一下。
- #work dir
- setwd('/lustre/user/liclab/sunym/R/rstudio/deseq2/')
- #load R package
- .libPaths('/lustre/user/liclab/sunym/R/rstudio/Rlib/')
- library(DESeq2)
- library(airway)
- library(tximeta)
- library(GEOquery)
- library(GenomicFeatures)
- library(BiocParallel)
- library(GenomicAlignments)
- #introduce of SummarizedExperiment using airway package
- data(gse)
载入airway中与整合好的gse数据,探索一下gse的特点。
- > gse
- class: RangedSummarizedExperiment
- dim: 58294 8
- metadata(6): tximetaInfo quantInfo ... txomeInfo txdbInfo
- assays(3): counts abundance length
- rownames(58294): ENSG00000000003.14 ENSG00000000005.5 ... ENSG00000285993.1
- ENSG00000285994.1
- rowData names(1): gene_id
- colnames(8): SRR1039508 SRR1039509 ... SRR1039520 SRR1039521
- colData names(3): names donor condition
- > assayNames(gse)
- [1] "counts" "abundance" "length"
- > head(assays(gse)[['counts']],3)
- SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517
- ENSG00000000003.14 708.164 467.962 900.992 424.368 1188.295 1090.668
- ENSG00000000005.5 0.000 0.000 0.000 0.000 0.000 0.000
- ENSG00000000419.12 455.000 510.000 604.000 352.000 583.000 773.999
- SRR1039520 SRR1039521
- ENSG00000000003.14 805.929 599.337
- ENSG00000000005.5 0.000 0.000
- ENSG00000000419.12 409.999 499.000
- > colSums(assay(gse))
- SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517 SRR1039520 SRR1039521
- 21100805 19298584 26145537 15688246 25268618 31891456 19683767 21813903
- > head(rowRanges(gse),3)
- GRanges object with 3 ranges and 1 metadata column:
- seqnames ranges strand | gene_id
- |
- ENSG00000000003.14 chrX 100627109-100639991 - | ENSG00000000003.14
- ENSG00000000005.5 chrX 100584802-100599885 | ENSG00000000005.5
- ENSG00000000419.12 chr20 50934867-50958555 - | ENSG00000000419.12
- -------
- seqinfo: 25 sequences (1 circular) from hg38 genome
- > seqinfo(rowRanges(gse))
- Seqinfo object with 25 sequences (1 circular) from hg38 genome:
- seqnames seqlengths isCircular genome
- chr1 248956422 FALSE hg38
- chr2 242193529 FALSE hg38
- chr3 198295559 FALSE hg38
- chr4 190214555 FALSE hg38
- chr5 181538259 FALSE hg38
- ... ... ... ...
- chr21 46709983 FALSE hg38
- chr22 50818468 FALSE hg38
- chrX 156040895 FALSE hg38
- chrY 57227415 FALSE hg38
- chrM 16569 TRUE hg38
- > colData(gse)
- DataFrame with 8 rows and 3 columns
- names donor condition
- SRR1039508 SRR1039508 N61311 Untreated
- SRR1039509 SRR1039509 N61311 Dexamethasone
- SRR1039512 SRR1039512 N052611 Untreated
- SRR1039513 SRR1039513 N052611 Dexamethasone
- SRR1039516 SRR1039516 N080611 Untreated
- SRR1039517 SRR1039517 N080611 Dexamethasone
- SRR1039520 SRR1039520 N061011 Untreated
- SRR1039521 SRR1039521 N061011 Dexamethasone
assay中可以存储不止一个matrix,assayNames函数可以列出所有的这些matrix,assay函数则默认返回assay中的第一个matrix。seqinfo函数可以获取基因组的相关信息。如果使用tximeta将测序转录本导入DESeq2,则通常会返回assay中的三个matrix:counts,各个基因与样本比对估计到的counts数;abundance,用TPM表示的基因丰度;length,基因的有效长度,来源于不同样本转录本的bias。
数据下载与比对
先建一个文本文档,输入全部的SRA号,方便我们后续批量处理数据。
- [email protected]:deseq2$more SRA.txt
- SRR1039508
- SRR1039509
- SRR1039512
- SRR1039513
- SRR1039516
- SRR1039517
- SRR1039520
- SRR1039521
使用ncbi提供的prefetch下载数据。
- nohup time /lustre/user/liclab/sunym/sratoolkit/sratoolkit.2.10.8-centos_linux64/bin/prefetch-orig.2.10.8 --option-file ./SRA.txt &
也可以调用ascp进行下载。
- #!/bin/bash
- /lustre/user/liclab/sunym/sratoolkit/sratoolkit.2.9.6-1-centos_linux64/bin/prefetch.2.9.3
- --ascp-path '/home/sunym/.aspera/connect/bin/ascp|/home/sunym/.aspera/connect/etc/asperaweb_id_dsa.openssh'
- --option-file ./SRA.txt
使用fasterq dump分解sra数据。
- for i in `cat ./SRA.txt`;do /lustre1/lch3000_pkuhpc/sunym/software/sratoolkit.2.10.0-centos_linux64/bin/fasterq-dump-orig.2.10.0 --split-3 -e 10 -O ./airway ./airway/$i.sra;done
STAR
使用STAR比对前需要先准备好STAR index,基因组注释文件以及参考基因组文件,基因组注释文件和参考基因组可以从GENCODE下载。构建STAR index代码如下。
- #!/bin/bash
- #SBATCH -J STAR
- #SBATCH -p fat4way
- #SBATCH -N 1
- #SBATCH -o STAR_%j.out
- #SBATCH -e STAR_%j.err
- #SBATCH --no-requeue
- #SBATCH -A lch3000_g1
- #SBATCH --qos=lch3000f4w
- #SBATCH -c 24
- pkurun /lustre1/lch3000_pkuhpc/sunym/envs/analyse/bin/STAR
- --runThreadN 20 --runMode genomeGenerate
- --genomeDir /lustre1/lch3000_pkuhpc/sunym/reference/GRCh38/star_index
- --genomeFastaFiles /lustre1/lch3000_pkuhpc/sunym/reference/GRCh38/genome/GRCh38.p13.genome.fa
- --sjdbGTFfile /lustre1/lch3000_pkuhpc/sunym/reference/GRCh38/annotation_gencode/gencode.v35.annotation.gtf
- --sjdbOverhang 100
STAR比对代码如下。
- #!/bin/bash
- #SBATCH -J STAR
- #SBATCH -p cn-short
- #SBATCH -N 1
- #SBATCH -o star_%j.out
- #SBATCH -e star_%j.err
- #SBATCH --no-requeue
- #SBATCH -A lch3000_g1
- #SBATCH --qos=lch3000cns
- #SBATCH -c 10
- pkurun /lustre1/lch3000_pkuhpc/sunym/envs/analyse/bin/STAR
- --runThreadN 10
- --genomeDir /lustre1/lch3000_pkuhpc/sunym/reference/UCSC/hg19/star_index
- --readFilesIn ./airway/SRR1039508.sra_1.fastq ./airway/SRR1039508.sra_2.fastq
- --outFileNamePrefix ./airway_star/SRR1039508.
STAR比对得到sam文件,使用samtools将其转换为bam文件。
- samtools view -bS -o aln.bam aln.sam
使用summarizeOverlaps函数生成SummarizedExperiment类。
- #work dir
- setwd('/lustre/user/liclab/sunym/R/rstudio/deseq2/')
- #load R package
- .libPaths('/lustre/user/liclab/sunym/R/rstudio/Rlib/')
- library(DESeq2)
- library(airway)
- library(tximeta)
- library(GEOquery)
- library(GenomicFeatures)
- library(BiocParallel)
- library(GenomicAlignments)
- #-----------Transcript quantification by STAR-----------------#
- remove(list = ls())
- #metadata
- dir <- system.file("extdata", package="airway", mustWork=TRUE)
- csvfile <- file.path(dir, "sample_table.csv")
- coldata <- read.csv(csvfile, row.names=1, stringsAsFactors=FALSE)
- coldata$names <- coldata$Run
- #生成bam文件list
- dir <- getwd()
- fls <- file.path(dir,'airway_bam',paste0(coldata$names,'.bam'))
- file.exists(fls)
- bamlist <- BamFileList(fls,yieldSize = 2000000)
- #生成feature
- txdb <- makeTxDbFromGFF('/lustre/user/liclab/sunym/reference/UCSC/hg19/annotation_gencode/gencode.v19.annotation.gtf')
- exonsByGene <- exonsBy(txdb,by='gene')
- #生成SE类
- register(MulticoreParam(workers = 8))
- SE <- summarizeOverlaps(features = exonsByGene,reads = bamlist,mode = 'Union',singleEnd = F,ignore.strand = T)
- #add coldata
- colnames(SE) <- sub(colnames(SE),pattern = '.bam',replacement = '')
- colnames(SE) == rownames(coldata)
- colData(SE) <- DataFrame(coldata)
这里为了方便直接从airway包中导出了样本的metadata;txdb类可以从gtf格式的基因组注释文件生成,包含了feature的信息;最后需要手动给SummarizedExperiment类加上sample的metadata(colData)。
Salmon
使用Salmon前需要准备好Salmon index,参考转录组和基因组注释文件;参考转录组和基因组注释文件可以从GENCODE网站下载。构建Salmon index代码如下:
- salmon index -t transcripts.fa.gz -i name_of_index
- #build index through gencode
- salmon index --gencode -t gencode.v29.transcripts.fa.gz -i gencode.v29_salmon_0.14.1
只需要指定转录组fasta文件位置以及指定的Salmon index输出文件夹名称即可,当然如果使用GENCODE下载的参考转录组,则可以加上相应的参数跳过一些多余的信息。Salmon比对代码如下:
- #!/bin/bash
- #SBATCH -J salmon
- #SBATCH -p cn-short
- #SBATCH -N 1
- #SBATCH -o salmon_%j.out
- #SBATCH -e salmon_%j.err
- #SBATCH --no-requeue
- #SBATCH -A lch3000_g1
- #SBATCH --qos=lch3000cns
- #SBATCH -c 10
- pkurun /lustre1/lch3000_pkuhpc/sunym/envs/analyse/bin/salmon quant
- -i /lustre1/lch3000_pkuhpc/sunym/reference/UCSC/hg19/Salmon_index/gencode.v34lift37_salmon_0.14.1
- -l A -p 10 --validateMappings --gcBias --numGibbsSamples 20
- -o ./salmon_quant/dirname
- -1 ./airway/SRR1039508.sra_1.fastq
- -2 ./airway/SRR1039508.sra_2.fastq
简单讲一下参数的意思,-i指定了我们所使用的Salmon index,-l A表示自动检测文库类型,-p 10表示使用10线程,–validateMappings表示使用可靠的mapping(默认参数),–gcBias矫正GC含量的系统偏差,最后三个参数则分别指定了比对后的输出路径以及双端测序的fastq文件路径。使用tximeta读取Salmon比对结果代码如下。
- #work dir
- setwd('/lustre/user/liclab/sunym/R/rstudio/deseq2/')
- #load R package
- .libPaths('/lustre/user/liclab/sunym/R/rstudio/Rlib/')
- library(DESeq2)
- library(airway)
- library(tximeta)
- library(GEOquery)
- library(GenomicFeatures)
- library(BiocParallel)
- library(GenomicAlignments)
- #-----------Transcript quantification by Salmon-----------------#
- remove(list = ls())
- #metadata
- dir <- system.file("extdata", package="airway", mustWork=TRUE)
- csvfile <- file.path(dir, "sample_table.csv")
- coldata <- read.csv(csvfile, row.names=1, stringsAsFactors=FALSE)
- coldata$names <- coldata$Run
- dir <- getwd()
- dir <- paste(dir,'airway_salmon',sep = '/')
- coldata$files <- file.path(dir,coldata$Run,'quant.sf')
- file.exists(coldata$files)
- #create SummarizedExperiment dataset
- makeLinkedTxome(indexDir='/lustre/user/liclab/sunym/R/rstudio/deseq2/Salmon_index/gencode.v35_salmon_0.14.1/', source="GENCODE", organism="Homo sapiens",
- release="35", genome="GRCh38", fasta='/lustre/user/liclab/sunym/R/rstudio/deseq2/transcript/gencode.v35.transcripts.fa', gtf='/lustre/user/liclab/sunym/R/rstudio/deseq2/annotation_gencode/gencode.v35.annotation.gtf', write=FALSE)
- se <- tximeta(coldata)
这里我们的metadata依旧从airway直接导出,tximeta的输入要求一张至少包含了names列和files列的数据表,names列指定了sample的名称,files列则指定了Salmon比对结果的完整路径。另外还需要在makeLinkedTxome中指定所使用的Salmon index、参考转录组以及基因组注释文件,这些文件与版本都要相互匹配。最后生成的SummarizedExperiment对象是以所有的转录本为行,而不同的转录本可能对应同一个基因,因此我们要根据makeLinkedTxome中设置的信息将转录本转换成基因。
- #explore se
- dim(se)
- head(rownames(se))
- gse <- summarizeToGene(se)
- dim(gse)
- head(rownames(gse))
简单看下输出。
- > #explore se
- > dim(se)
- [1] 228754 8
- > head(rownames(se))
- [1] "ENST00000456328.2" "ENST00000450305.2" "ENST00000488147.1" "ENST00000619216.1"
- [5] "ENST00000473358.1" "ENST00000469289.1"
- > gse <- summarizeToGene(se)
- loading existing TxDb created: 2020-08-22 17:46:33
- obtaining transcript-to-gene mapping from TxDb
- loading existing gene ranges created: 2020-08-22 17:49:16
- summarizing abundance
- summarizing counts
- summarizing length
- summarizing inferential replicates
- > dim(gse)
- [1] 60237 8
- > head(rownames(gse))
- [1] "ENSG00000000003.15" "ENSG00000000005.6" "ENSG00000000419.13" "ENSG00000000457.14"
- [5] "ENSG00000000460.17" "ENSG00000000938.13"
可以看一下转录组和基因组的命名,ENS意思是“ENSENMBLE”,表示使用ENSENMBLE ID进行命名,T表示转录组,G表示基因组。
到目前为止,我给出了GEO下载、STAR和Salmon比对,以及将比对文件(.bam,.sf)读入bioconductor的SummarizedExperiment通用类的代码。接下来即可将SummarizedExperiment类输入DESeq2构建成DESeqDataSet对象。下面提供了两种构建DESeqDataSet对象的方法,一种是直接从SummarizedExperiment转化而来,另一种是从count matrix和metadata从头构建。
构建DESeqDataSet对象
DeSeq2的主要目标就是做转录组基因差异表达分析,因此我们需要告诉计算机我们的分组信息以及用什么组与什么组进行比较,从而得到差异表达基因。DESeq2的差异表达分析的设计需要在一开始就使用design formula告诉DESeq2,最简单的design formula是~ condition,condition是colData中的一个列名,那么这样一个design formula就告诉DESeq2按照condition对不同的sample进行分组准备后续的数据处理和差异表达分析。在本实验数据中,我们使用~ cell dex,表示我们希望在控制细胞系的情况下,探究地塞米松处理的影响与差异表达情况。更复杂的design formula,如~ group treatment group:treatment,表示我们希望在控制不同的group和treatment的同时,探究不同group和treatment组合的影响(determine for which genes the effect of treatment is different across groups)。
从SummarizedExperiment对象构建
直接使用Bioconductor维护的通用对象构建DESeqDataSet对象相对比较简单,只需要设置对应的design formula即可,代码如下。
- #DESeqDataSet
- #start from SummarizedExperiment set
- round( colSums(assay(SE))/1e6, 1 )
- colData(SE)
- SE$dex
- dds <- DESeqDataSet(SE, design = ~ cell dex)
看下输出。
- > #start from SummarizedExperiment set
- > round( colSums(assay(se))/1e6, 1 )
- SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517 SRR1039520 SRR1039521
- 21.1 19.3 26.2 15.7 25.3 32.0 19.7 21.9
- > colData(se)
- DataFrame with 8 rows and 10 columns
- SampleName cell dex albut Run avgLength
- SRR1039508 GSM1275862 N61311 untrt untrt SRR1039508 126
- SRR1039509 GSM1275863 N61311 trt untrt SRR1039509 126
- SRR1039512 GSM1275866 N052611 untrt untrt SRR1039512 126
- SRR1039513 GSM1275867 N052611 trt untrt SRR1039513 87
- SRR1039516 GSM1275870 N080611 untrt untrt SRR1039516 120
- SRR1039517 GSM1275871 N080611 trt untrt SRR1039517 126
- SRR1039520 GSM1275874 N061011 untrt untrt SRR1039520 101
- SRR1039521 GSM1275875 N061011 trt untrt SRR1039521 98
- Experiment Sample BioSample names
- SRR1039508 SRX384345 SRS508568 SAMN02422669 SRR1039508
- SRR1039509 SRX384346 SRS508567 SAMN02422675 SRR1039509
- SRR1039512 SRX384349 SRS508571 SAMN02422678 SRR1039512
- SRR1039513 SRX384350 SRS508572 SAMN02422670 SRR1039513
- SRR1039516 SRX384353 SRS508575 SAMN02422682 SRR1039516
- SRR1039517 SRX384354 SRS508576 SAMN02422673 SRR1039517
- SRR1039520 SRX384357 SRS508579 SAMN02422683 SRR1039520
- SRR1039521 SRX384358 SRS508580 SAMN02422677 SRR1039521
- > se$dex
- [1] "untrt" "trt" "untrt" "trt" "untrt" "trt" "untrt" "trt"
- > dds <- DESeqDataSet(se, design = ~ cell dex)
- using counts and average transcript lengths from tximeta
- Warning message:
- In DESeqDataSet(se, design = ~cell dex) :
- some variables in design formula are characters, converting to factors
- >
其实只需要最后一行代码调用DESeqDataSet函数,设定design formula即可。
- #更改因子水平
- dds$dex
- dds$dex <- factor(dds$dex,levels = c('untrt','trt'))
- dds$dex
看下输出。
- > dds$dex
- [1] untrt trt untrt trt untrt trt untrt trt
- Levels: trt untrt
- > dds$dex <- factor(dds$dex,levels = c('untrt','trt'))
- > dds$dex
- [1] untrt trt untrt trt untrt trt untrt trt
- Levels: untrt trt
- >
可以看到DESeqDataSet对象的dex metadata有两个水平,一个是经过dex处理,一个是未经过dex处理。在R中,通常control组(未处理)放在前面,因此上面的代码实现的就是把因子水平的顺序换了一下。到这里,dds这一DESeqDataSet对象就可以用于DESeq2后续的分析了。
从count table构建
事实上DESeqDataSet对象也就只包含三部分内容:count table,metadata以及design formula。因此我们也可以直接从count table进行构建。
- #start from count matrix
- countdata <- round(assay(SE))
- head(countdata, 3)
- coldata <- colData(SE)
- dds <- DESeqDataSetFromMatrix(countData = countdata,
- colData = coldata,
- design = ~ cell dex)
- dds$dex
- dds$dex <- factor(dds$dex,levels = c('untrt','trt'))
上面的演示从之前的SE对象中获取count table和metadata进行构建。
OK,DESeq2的分析前准备到这里就结束了,最后老规矩,贴一个R中的完整代码。
- #sleep
- ii <- 1
- while(1){
- cat(paste("round",ii),sep = "n")
- ii <- ii 1
- Sys.sleep(30)
- }
- #work dir
- setwd('/lustre/user/liclab/sunym/R/rstudio/deseq2/')
- #load R package
- .libPaths('/lustre/user/liclab/sunym/R/rstudio/Rlib/')
- library(DESeq2)
- library(airway)
- library(tximeta)
- library(GEOquery)
- library(GenomicFeatures)
- library(BiocParallel)
- library(GenomicAlignments)
- #introduce of SummarizedExperiment using airway package
- data(gse)
- gse
- assayNames(gse)
- head(assays(gse)[['counts']],3)
- colSums(assay(gse))
- head(rowRanges(gse),3)
- seqinfo(rowRanges(gse))
- colData(gse)
- #-----------Transcript quantification by Salmon-----------------#
- remove(list = ls())
- #metadata
- dir <- system.file("extdata", package="airway", mustWork=TRUE)
- csvfile <- file.path(dir, "sample_table.csv")
- coldata <- read.csv(csvfile, row.names=1, stringsAsFactors=FALSE)
- coldata$names <- coldata$Run
- dir <- getwd()
- dir <- paste(dir,'airway_salmon',sep = '/')
- coldata$files <- file.path(dir,coldata$Run,'quant.sf')
- file.exists(coldata$files)
- #create SummarizedExperiment dataset
- makeLinkedTxome(indexDir='/lustre/user/liclab/sunym/R/rstudio/deseq2/Salmon_index/gencode.v35_salmon_0.14.1/', source="GENCODE", organism="Homo sapiens",
- release="35", genome="GRCh38", fasta='/lustre/user/liclab/sunym/R/rstudio/deseq2/transcript/gencode.v35.transcripts.fa', gtf='/lustre/user/liclab/sunym/R/rstudio/deseq2/annotation_gencode/gencode.v35.annotation.gtf', write=FALSE)
- se <- tximeta(coldata)
- #explore se
- dim(se)
- head(rownames(se))
- gse <- summarizeToGene(se)
- dim(gse)
- head(rownames(gse))
- #-----------Transcript quantification by STAR-----------------#
- remove(list = ls())
- #metadata
- dir <- system.file("extdata", package="airway", mustWork=TRUE)
- csvfile <- file.path(dir, "sample_table.csv")
- coldata <- read.csv(csvfile, row.names=1, stringsAsFactors=FALSE)
- coldata$names <- coldata$Run
- #生成bam文件list
- dir <- getwd()
- fls <- file.path(dir,'airway_bam',paste0(coldata$names,'.bam'))
- file.exists(fls)
- bamlist <- BamFileList(fls,yieldSize = 2000000)
- #生成feature
- txdb <- makeTxDbFromGFF('/lustre/user/liclab/sunym/reference/UCSC/hg19/annotation_gencode/gencode.v19.annotation.gtf')
- exonsByGene <- exonsBy(txdb,by='gene')
- #生成SE类
- register(MulticoreParam(workers = 8))
- SE <- summarizeOverlaps(features = exonsByGene,reads = bamlist,mode = 'Union',singleEnd = F,ignore.strand = T)
- #add coldata
- colnames(SE) <- sub(colnames(SE),pattern = '.bam',replacement = '')
- colnames(SE) == rownames(coldata)
- colData(SE) <- DataFrame(coldata)
- #------------------------------------------------------------------#
- #------------------------------------------------------------------#
- #DESeqDataSet
- #start from SummarizedExperiment set
- round( colSums(assay(se))/1e6, 1 )
- colData(se)
- se$dex
- dds <- DESeqDataSet(se, design = ~ cell dex)
- dds$dex
- dds$dex <- factor(dds$dex,levels = c('untrt','trt'))
- dds$dex
- #start from count matrix
- countdata <- round(assay(SE))
- head(countdata, 3)
- coldata <- colData(SE)
- dds <- DESeqDataSetFromMatrix(countData = countdata,
- colData = coldata,
- design = ~ cell dex)
- dds$dex
- dds$dex <- factor(dds$dex,levels = c('untrt','trt'))