

除了整合差异相对较小的scRNA-seq数据集外,liger也能用于整合数据差异较大、甚至研究的对象完全不同的多组学。本文将以scRNA-seq和scATAC-seq为例,展示liger多组学整合的相关流程。scRNA-seq描述了细胞的转录组特征,而scATAC-seq则描述了细胞基因组开放性的特征,显然这两种组学特征之间存在着某种相关性,大胆一点我们可以认为整体上呈现正相关。通过scRNA-seq和scATAT-seq的整合分析,我们不光能够更加全面地描述细胞的特征,也能从开放性和RNA的角度上去推测可能的基因表达调控模式。尽管现在已经有了一些多组学测序的技术,例如10x的Single Cell Multiome ATAC Gene Expression,但更多的组学特征以及技术上仍有待进一步的突破,因此liger依然给我们提供了一个非常好的窗口帮助我们理解单细胞的生命特征。
对不同的数据集进行整合,数据集之间需要有一些共有的对象来作为feature,并且这些feature在不同的数据集之间应当存在着某种整体上的相关性(例如某一个基因越开放,则通常它的表达量就越高;某一个基因body上非CpG甲基化程度越高,则该基因通常就越处于沉默状态)。对于ATAC-seq等表观组的数据,我们通常使用peak来表征其在某一个位点上的强弱。尽管我们也可以统计各个基因上的peak数量来作为ATAC-seq的表达矩阵,但liger的作者认为这么做的效果可能并不理想,原因有以下三点。
- Call peak通常是用全部细胞来做的,而这种方式很容易埋没一些稀有细胞群的信号。
- 基因body上的开放性往往是比较弥散的,不像一些调控元件可以call出非常明显的peak,因此peaking calling的算法可能会丢失大量的信息。
- Peak本身就类似于显著差异的变化,因此大量的信息被丢失,本身稀疏的数据变得更加稀疏。
Liger作者发现,使用scATAC-seq在基因body和promoter上的reads数就能很容易地表征某个基因的整体开放性水平,promoter区域可以选择TSS上下游2kb的区域。本文的分析使用10x提供的公共测试数据,并使用10x提供的cellranger和cellranger-atac进行比对。scRNA-seq数据为健康供体的10k PBMC数据,scATAC-seq数据也是健康供体的10k PBMC数据。scATAC-seq比对所用的reference来自直接从10x官网上下载的现成的GRCh38参考基因组与注释,该reference所用的基因组注释文件为GENCODE的第28版GRCh38基因组注释,下载该注释文件后我们手工构建一个scRNA-seq的reference,从而最大程度地保证了scRNA-seq和scATAC-seq所用的注释文件的一致性。
- ############################################
- ## Project: Liger-learning
- ## Script Purpose: joint analysis of scRNA-seq and chromatin accessibility
- ## Data: 2020.10.24
- ## Author: Yiming Sun
- ############################################
- #sleep
- ii <- 1
- while(1){
- cat(paste("round",ii),sep = "n")
- ii <- ii 1
- Sys.sleep(30)
- }
- # general setting
- setwd('/data/User/sunym/project/liger_learning/')
- # library
- library(Seurat)
- library(ggplot2)
- library(dplyr)
- library(scibet)
- library(Matrix)
- library(tidyverse)
- library(cowplot)
- library(viridis)
- library(pheatmap)
- library(ComplexHeatmap)
- library(networkD3)
- library(liger)
- library(parallel)
- #my function
- MySuggestLambda <- function(object,lambda,k,knn_k = 20,thresh = 1e-04,max.iters = 100,k2 = 500,ref_dataset = NULL,resolution = 1,parl_num){
- res <- matrix(ncol = 2,nrow = length(lambda))
- colnames(res) <- c('lambda','alignment')
- res[,'lambda'] <- lambda
- cl <- makeCluster(parl_num)
- clusterExport(cl,c('object','k','knn_k','thresh','max.iters','k2','ref_dataset','resolution'),envir = environment())
- clusterExport(cl,c('optimizeALS','quantileAlignSNF','calcAlignment'),envir = .GlobalEnv)
- alignment_score <- parLapply(cl = cl,lambda,function(x){
- object <- optimizeALS(object,k = k,lambda = x,thresh = thresh,max.iters = max.iters)
- object <- quantileAlignSNF(object,knn_k = knn_k,k2 = k2,ref_dataset = ref_dataset,resolution = resolution)
- return(calcAlignment(object))
- })
- stopCluster(cl)
- res[,'alignment'] <- (alignment_score %>% unlist())
- res <- as.data.frame(res)
- res[,1] <- as.numeric(as.character(res[,1]))
- res[,2] <- as.numeric(as.character(res[,2]))
- p <- ggplot(res,aes(x = lambda,y = alignment)) geom_point() geom_line()
- return(list(res,p))
- }
scATAC-seq 数据准备
首先我们需要把GENCODE提供的gtf格式的基因组注释文件转换成bed格式的注释文件,再用bedmap等工具去寻找我们测序所得到的reads与基因组上的注释有哪些overlap。gtf文件的前五行为该注释文件的相关信息,需要先去掉。
- [[email protected] annotation]$ head -n 5 gencode.v28.annotation.gtf
- ##description: evidence-based annotation of the human genome (GRCh38), version 28 (Ensembl 92)
- ##provider: GENCODE
- ##contact: [email protected]
- ##format: gtf
- ##date: 2018-03-23
为了方便写成脚本,我就直接在R中调用system函数了,实际过程中我们也可以直接在linux终端进行操作。
- # 1.joint analysis of scRNA-seq and chromatin accessibility
- # pre-prapare scTATA data
- #sort by chromosome start position and end position for fragment, gene and promoter
- system('gunzip ./data/10k_pbmc/scATAC/fragments.tsv.gz')
- system('tail -n 6 ./data/10k_pbmc/annotation/gencode.v28.annotation.gtf > ./data/10k_pbmc/annotation/genes_split.gtf')
- genes <- read.table('./data/10k_pbmc/annotation/genes_split.gtf',sep = 't')
读入gtf文件后我们需要对条目进行筛选和整理。V1列表示所在的染色体,V4和V5分别表示了该片段在染色体上的起始位置,V7表示该片段位于正义链还是反义链,V9列则是对该片段名称的注释。我们筛选V3列为gene的条目,并对V9列进行整理。
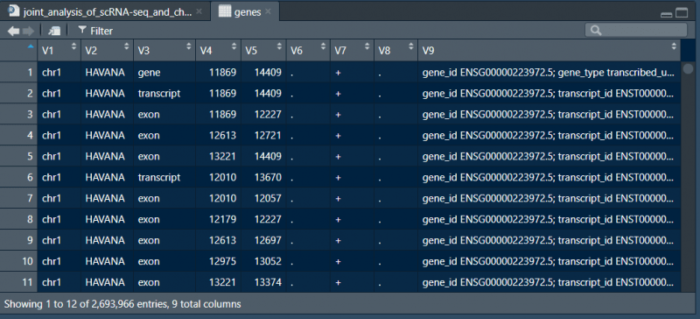
- genes <- genes[genes[,3] == 'gene',]
- gene_name <- unlist(strsplit(as.character(genes[,9]),split = '; ',fixed = TRUE))
- gene_name <- gene_name[grepl(pattern = 'gene_name',gene_name)]
- gene_name <- sub(pattern = 'gene_name ',replacement = '',gene_name)
- genes[,9] <- gene_name
- genes <- genes[,c(1,4,5,9,6,7)]
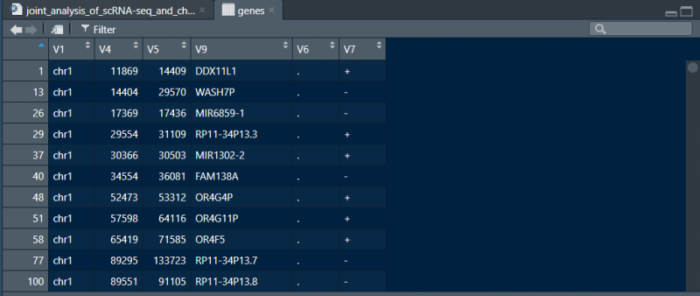
这样我们就得到了bed类似格式的注释文件。同理我们也可以利用TSS得到promoter的位置信息,不过需要注意正义链和反义链。构建好基因和promoter的两个表格后,将其保存为bed文件即可。
- promoter <- as.data.frame(matrix(nrow = dim(genes)[1],ncol = dim(genes)[2]))
- promoter[,c(1,4,5,6)] <- genes[,c(1,4,5,6)]
- for (i in 1:dim(promoter)[1]) {
- if(promoter[i,6] == ' '){
- if ((as.numeric(genes[i,2])-2000) >= 0){
- promoter[i,2] <- as.numeric(genes[i,2])-2000
- }else{
- promoter[i,2] <- 0
- }
- promoter[i,3] <- as.numeric(genes[i,2]) 2000
- } else if(promoter[i,6] == '-'){
- if ((as.numeric(genes[i,3])-2000) >= 0){
- promoter[i,2] <- as.numeric(genes[i,3])-2000
- }else{
- promoter[i,2] <- 0
- }
- promoter[i,3] <- as.numeric(genes[i,3]) 2000
- }
- }
- write.table(genes,file = './data/10k_pbmc/annotation/genes.bed',sep = 't',col.names = FALSE,row.names = FALSE,quote = FALSE)
- write.table(promoter,file = './data/10k_pbmc/annotation/promoter.bed',sep = 't',col.names = FALSE,row.names = FALSE,quote = FALSE)
在用bedmap做overlap之前需要先对reads和bed文件进行排序,按照染色体、起始位置、结束位置三者的先后顺序进行排序。排序完成后即可用bedmap计算overlap。
- system('/data/User/sunym/software/BEDOPS/bin/sort-bed --max-mem 50G ./data/10k_pbmc/scATAC/fragments.tsv > ./data/10k_pbmc/scATAC/atac_fragments.sort.bed')
- system('/data/User/sunym/software/BEDOPS/bin/sort-bed --max-mem 50G ./data/10k_pbmc/annotation/genes.bed > ./data/10k_pbmc/annotation/genes.sort.bed')
- system('/data/User/sunym/software/BEDOPS/bin/sort-bed --max-mem 50G ./data/10k_pbmc/annotation/promoter.bed > ./data/10k_pbmc/annotation/promoters.sort.bed')
- #Use bedmap command to calculate overlapping elements between indexes and fragment output files
- system('/data/User/sunym/software/BEDOPS/bin/bedmap --ec --delim "t" --echo --echo-map-id ./data/10k_pbmc/annotation/promoters.sort.bed ./data/10k_pbmc/scATAC/atac_fragments.sort.bed > ./data/10k_pbmc/scATAC/atac_promoters_bc.bed')
- system('/data/User/sunym/software/BEDOPS/bin/bedmap --ec --delim "t" --echo --echo-map-id ./data/10k_pbmc/annotation/genes.sort.bed ./data/10k_pbmc/scATAC/atac_fragments.sort.bed > ./data/10k_pbmc/scATAC/atac_genes_bc.bed')
构建liger对象
读入bedmap计算overlap后的输出文件。
- #import the bedmap outputs into the R
- genes.bc <- read.table(file = "./data/10k_pbmc/scATAC/atac_genes_bc.bed", sep = "t", as.is = c(4,7), header = FALSE)
- promoters.bc <- read.table(file = "./data/10k_pbmc/scATAC/atac_promoters_bc.bed", sep = "t", as.is = c(4,7), header = FALSE)
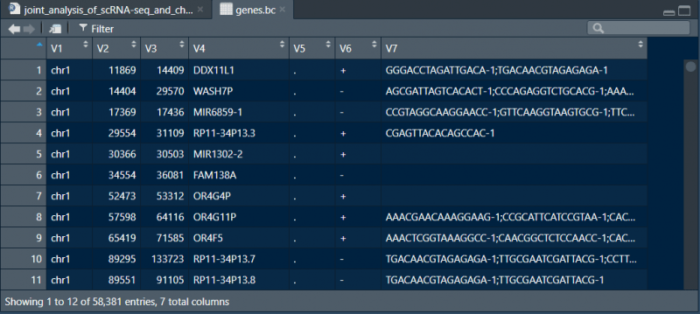
可以看到V7列包括了所有与该基因有overlap的细胞的barcode。筛选至少覆盖1500个基因的细胞为有效的细胞,并与peak数据中过滤得到的barcode做overlap,将这部分细胞用于后续的分析。
- #load scATAC peak filted barcode data
- barcodes <- read.table(file = './data/10k_pbmc/scATAC/filtered_peak_bc_matrix/barcodes.tsv',sep = 't',as.is = TRUE,header = FALSE)$V1
- #filter the barcode with bad quality
- bc <- genes.bc[,7]
- bc_split <- strsplit(bc,";")
- bc_split_vec <- unlist(bc_split)
- bc_counts <- table(bc_split_vec)
- bc_filt <- names(bc_counts)[bc_counts > 1500]
- barcodes <- intersect(bc_filt,barcodes)
使用liger自带的makeFeatureMatrix函数对bedmap的输出结果进行处理,得到单个细胞在各个基因及其promoter上的reads数的表达矩阵,用该矩阵和scRNA-seq表达矩阵构建liger对象。
- #use LIGER's makeFeatureMatrix function to calculate accessibility counts for gene body and promoter
- gene.counts <- makeFeatureMatrix(genes.bc, barcodes)
- promoter.counts <- makeFeatureMatrix(promoters.bc, barcodes)
- gene.counts <- gene.counts[order(rownames(gene.counts)),]
- promoter.counts <- promoter.counts[order(rownames(promoter.counts)),]
- PBMCs <- gene.counts promoter.counts
- colnames(PBMCs)=paste0("PBMC_",colnames(PBMCs))
- #load scRNA-seq data
- PBMC.rna <- read10X(sample.dirs = './data/10k_pbmc/scRNA/filtered_feature_bc_matrix/',sample.names = 'rna')
- #create a LIGER object
- PBMC.data <- list(atac = PBMCs, rna = PBMC.rna)
- int.PBMC <- createLiger(PBMC.data)
- remove(list = ls()[ls() != 'int.PBMC'])
- gc()
对liger对象进行标准化等一系列前期操作,注意只使用RNA-seq数据来挑选variable gene。
- #normalize select gene and scale
- int.PBMC <- normalize(int.PBMC)
- int.PBMC <- selectGenes(int.PBMC, datasets.use = 2)
- int.PBMC <- scaleNotCenter(int.PBMC)
非负矩阵分解
选择合适的参数K和λ进行矩阵分解。
- #integrate NMF
- #suggest lambda
- suggestLambda(int.PBMC,lambda.test = seq(1,20,1),k = 25,num.cores = 10)
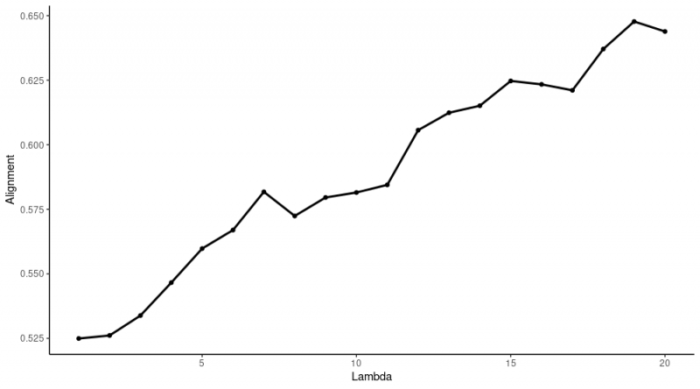
看这趋势似乎还在一路走高,一般建议λ的取值范围为0.25到60。经过后续的降维,我发现λ=10是个不错的选择。
- #suggest k
- suggestK(object = int.PBMC,k.test = seq(10,30,1),lambda = 10,num.cores = 10,plot.log2 = FALSE)
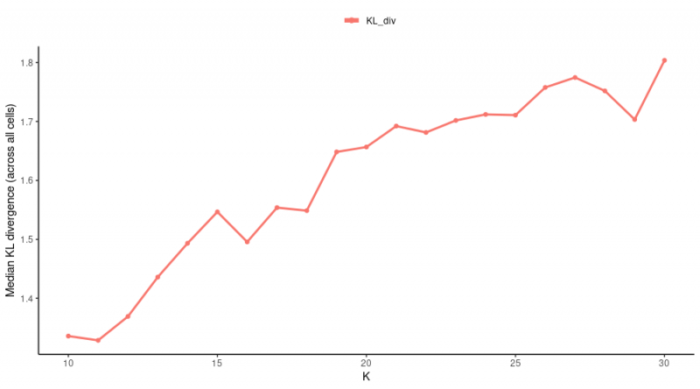
K在25左右就基本达到了饱和,因此我们选择K=25。根据我们后续的降维和聚类,这些值也都可以人为的进行调整,使得它尽可能地符合你的数据集,并让你的结果看起来“干净”。
- #lambda = 10,k = 25
- int.PBMC <- optimizeALS(int.PBMC,k = 25,lambda = 10,max.iters = 30,thresh = 1e-06)
- #Quantile Normalization and Joint Clustering
- int.PBMC <- quantile_norm(int.PBMC,knn_k = 20,quantiles = 50,min_cells = 20,do.center = FALSE,
- max_sample = 1000,refine.knn = TRUE,eps = 0.9)
- int.PBMC <- louvainCluster(int.PBMC, resolution = 0.4)
可以看下总共聚出了多少个cluster。
- > table([email protected])
- 0 1 2 3 4 5 6 7 8 9 10 11
- 3360 2552 2484 2074 1976 1683 1629 1004 897 778 776 416
- 12 13 14 15 16 17
- 398 391 316 304 154 108
可视化
- UMAP
- #Visualization and Downstream Analysis
- int.PBMC <- runUMAP(int.PBMC)
- all.plots <- plotByDatasetAndCluster(int.PBMC, axis.labels = c('UMAP 1', 'UMAP 2'),return.plots = TRUE)
- head(all.plots[[1]]$data)
- pdf(file = './res/fig_201112/plot_by_dataset_and_cluster.pdf',width = 8,height = 4)
- all.plots[[1]] all.plots[[2]]
- dev.off()
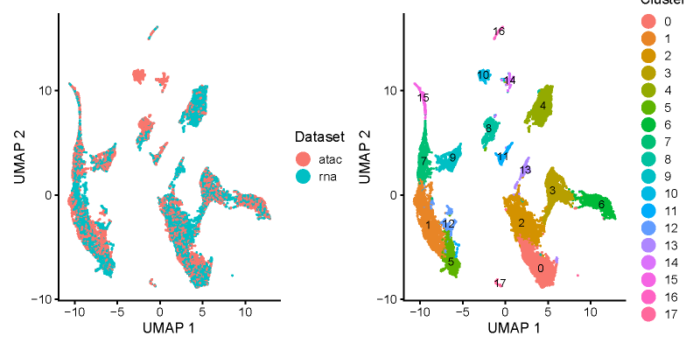
- 差异表达分析
- #identify marker genes for each cluster
- int.PBMC.wilcoxon <- runWilcoxon(int.PBMC, data.use = 'all', compare.method = 'clusters')
- #filter the enriched gene yourself
- int.PBMC.wilcoxon <- int.PBMC.wilcoxon[int.PBMC.wilcoxon$padj < 0.05,]
- int.PBMC.wilcoxon <- int.PBMC.wilcoxon[int.PBMC.wilcoxon$logFC > 3,]
- #top 20 markers of cluster 3 and 4
- wilcoxon.cluster_3 <- int.PBMC.wilcoxon[int.PBMC.wilcoxon$group == 3, ]
- wilcoxon.cluster_3 <- wilcoxon.cluster_3[order(wilcoxon.cluster_3$padj), ]
- markers.cluster_3 <- wilcoxon.cluster_3[1:20, ]
- wilcoxon.cluster_4 <- int.PBMC.wilcoxon[int.PBMC.wilcoxon$group == 4, ]
- wilcoxon.cluster_4 <- wilcoxon.cluster_4[order(wilcoxon.cluster_4$padj), ]
- markers.cluster_4 <- wilcoxon.cluster_4[1:20, ]
具体看下cluster_3和cluster_4中有哪些基因(RNA和开放性)被富集到。
- > markers.cluster_3
- feature group avgExpr logFC statistic auc pval padj pct_in pct_out
- 93440 AC092580.4 3 -16.77217 5.369608 27507364 0.6898446 0 0 100 100
- 96414 CCL5 3 -11.02601 9.269639 32396409 0.8124547 0 0 100 100
- 96562 CD8A 3 -13.57582 6.735737 29898397 0.7498083 0 0 100 100
- 97532 CST7 3 -12.67376 7.845358 31058233 0.7788952 0 0 100 100
- 98887 CTSW 3 -14.43134 6.331951 28632810 0.7180692 0 0 100 100
- 99965 EOMES 3 -16.80980 4.705106 26695598 0.6694867 0 0 100 100
- 101646 GZMA 3 -14.08326 7.198335 29369425 0.7365424 0 0 100 100
- 101649 GZMK 3 -14.48547 7.763756 30122792 0.7554357 0 0 100 100
- 101650 GZMM 3 -13.68788 6.104824 28699408 0.7197393 0 0 100 100
- 102068 HOPX 3 -15.09229 5.371568 27605779 0.6923127 0 0 100 100
- 102524 IL32 3 -11.36195 6.950100 29875540 0.7492350 0 0 100 100
- 103205 KLRB1 3 -14.85939 6.264977 28100056 0.7047085 0 0 100 100
- 103213 KLRG1 3 -11.01856 7.183600 31721918 0.7955395 0 0 100 100
- 103361 LAG3 3 -18.79355 3.451362 24980309 0.6264698 0 0 100 100
- 103916 LYAR 3 -12.56974 5.956998 29655544 0.7437179 0 0 100 100
- 104130 MATK 3 -15.48198 5.632058 28084886 0.7043280 0 0 100 100
- 105193 NCR3 3 -17.23250 4.670144 26521096 0.6651105 0 0 100 100
- 105421 NKG7 3 -11.84455 8.689092 31538326 0.7909353 0 0 100 100
- 107166 PRF1 3 -13.84797 7.254678 30034053 0.7532103 0 0 100 100
- 107282 PRR5 3 -12.95011 4.999333 28880551 0.7242821 0 0 100 100
- > markers.cluster_4
- feature group avgExpr logFC statistic auc pval padj pct_in pct_out
- 124628 ADAM28 4 -14.686500 6.687493 28617138 0.7494493 0 0 100 100
- 125560 ARHGAP24 4 -13.124318 5.737005 28321370 0.7417034 0 0 100 100
- 126016 BANK1 4 -9.488431 9.653939 34707167 0.9089400 0 0 100 100
- 126063 BCL11A 4 -11.083597 7.253749 31052386 0.8132255 0 0 100 100
- 126149 BLK 4 -13.199351 7.485614 30191285 0.7906743 0 0 100 100
- 126152 BLNK 4 -16.619583 4.727523 25910558 0.6785671 0 0 100 100
- 127204 CD22 4 -11.490107 9.400583 32656222 0.8552281 0 0 100 100
- 127206 CD24 4 -17.198765 5.403821 26269448 0.6879660 0 0 100 100
- 127229 CD37 4 -7.768716 6.736971 33587469 0.8796164 0 0 100 100
- 127236 CD40 4 -15.849347 5.536785 27024648 0.7077438 0 0 100 100
- 127244 CD52 4 -9.103628 5.641797 30316353 0.7939497 0 0 100 100
- 127257 CD74 4 -6.196057 6.340909 34263853 0.8973301 0 0 100 100
- 127258 CD79A 4 -8.407126 12.678446 35987290 0.9424649 0 0 100 100
- 127259 CD79B 4 -9.626859 11.530288 34664105 0.9078122 0 0 100 100
- 127313 CDCA7L 4 -16.539789 4.886703 26014286 0.6812836 0 0 100 100
- 127941 COBLL1 4 -16.594804 4.437667 25557096 0.6693103 0 0 100 100
- 130946 FAM129C 4 -13.668283 6.205986 28893095 0.7566762 0 0 100 100
- 131192 FAU 4 -8.118877 4.119389 29235631 0.7656468 0 0 100 100
- 131273 FCER2 4 -14.723589 6.953663 28740433 0.7526782 0 0 100 100
- 131280 FCGR2B 4 -19.050128 3.596260 23898797 0.6258814 0 0 100 100
- >
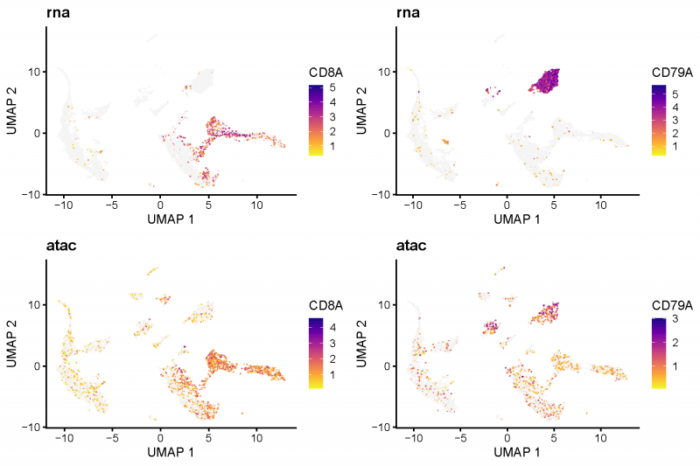
cluster_3中富集到了CD8A,cluster_4中富集到了CD79A,感觉都是挺重要的基因,可以看下这两个基因的表达情况。
- #feature plot on umap
- # CD8A for cluster 2 and CD79A for cluster 4
- CD8A <- plotGene(int.PBMC, "CD8A", axis.labels = c('UMAP 1', 'UMAP 2'), return.plots = TRUE)
- CD79A <- plotGene(int.PBMC, "CD79A", axis.labels = c('UMAP 1', 'UMAP 2'), return.plots = TRUE)
- pdf(file = './res/fig_201112/marker_for_cluster_3_and_4.pdf',width = 9,height = 6)
- plot_grid(CD8A[[2]],CD79A[[2]],CD8A[[1]],CD79A[[1]], ncol=2)
- dev.off()
- metagene
也可以从metagene的角度去探究基因的差异表达。
- #gene loading plot
- # gene loading plot of factor12
- all.plots <- plotGeneLoadings(int.PBMC, return.plots = TRUE,do.spec.plot = FALSE)
- pdf(file = './res/fig_201112/gene_loading_factor_18.pdf')
- all.plots[[18]]
- dev.off()
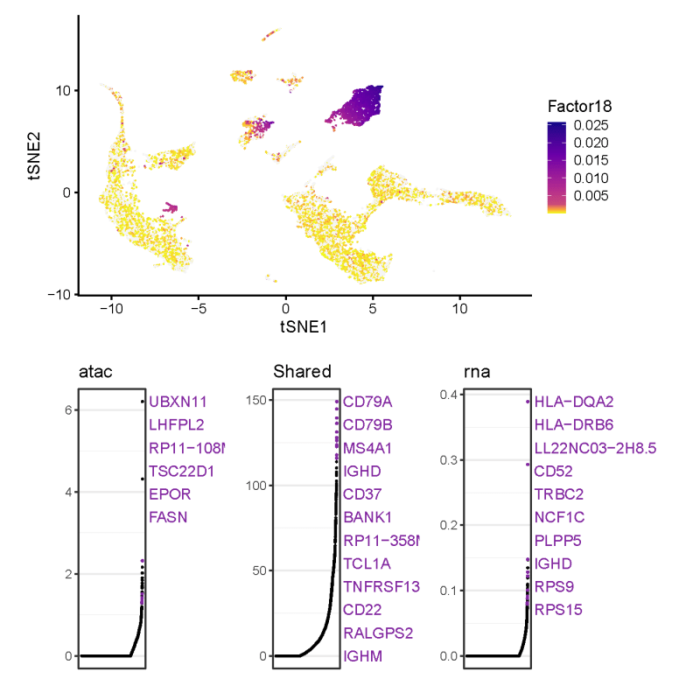
可以看到metagene_18在cluster_4中特异性地高loading,两个数据集中CD79A和CD79B都对metagene_18有很高的贡献,这与我们之前找到的差异表达基因相吻合。可以继续来看下不同数据集各自对metagene_18的贡献,如何导出gene loading plot背后的数据可以参考文章使用liger整合单细胞RNA-seq数据集。
- # feature plot
- TCL1A <- plotGene(int.PBMC, "TCL1A", axis.labels = c('UMAP 1', 'UMAP 2'), return.plots = TRUE)
- RPS9 <- plotGene(int.PBMC, "RPS9", axis.labels = c('UMAP 1', 'UMAP 2'), return.plots = TRUE)
- pdf(file = './res/fig_201112/RPS9_TCL1A_plot_for_RNA_and_ATAC.pdf',width = 9,height = 6)
- plot_grid(RPS9[[2]],TCL1A[[2]],RPS9[[1]],TCL1A[[1]], ncol=2)
- dev.off()
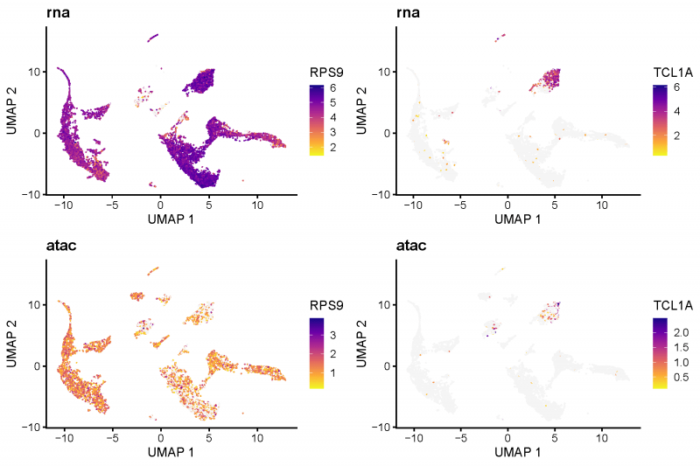
可以看到RPS9作为一种核糖体蛋白在各种细胞中均有表达,而在分泌抗体的B细胞中这种表达显然会更高,这与RNA-seq的表达谱相一致。不过在ATAC-seq的表达谱中,B细胞中的RPS9的开放性似乎并不是最高的,说明控制RPS9转录的机制可能不止基因组的开放性这一种。TCL1A的转录组和ATAC-seq谱在都在B细胞中高表达,显示出了开放性与基因表达的整体正相关。
- Pseudo peak
这也是liger非常有意思的一点,即它可以利用一个数据集的特征去预测另一个数据集的特征。想法也非常简单,数据集1中的细胞A周围有k个数据集2中的细胞,那么A的特征应该和这k个细胞很像,利用简单的平均我们就可以预测出A中数据集2特有的特征。在本例中,ATAC-seq的peak是数据集2(ATAC-seq数据集)特有的特征,那么我可以利用liger预测出数据集1(RNA-seq数据集)中细胞的peak特征,就像我们同时对一个细胞测定了RNA和ATAC一样,尽管这种ATAC是预测出来的。
首先我们需要先读入ATAC-seq的peak的数据,并对barcode进行过滤,使得它完全包含于我们之前使用reads构建的ATAC-seq表达谱中。
- # Pseudo single multi-omic data (scRNA-seq and scATAC-seq)
- #load scATAC peak data
- barcodes <- read.table(file = './data/10k_pbmc/scATAC/filtered_peak_bc_matrix/barcodes.tsv',sep = 't',as.is = TRUE,header = FALSE)$V1
- barcodes <- paste('PBMC',barcodes,sep = '_')
- peak.names <- read.table(file = './data/10k_pbmc/scATAC/filtered_peak_bc_matrix/peaks.bed', sep = 't', header = FALSE)
- peak.names <- paste0(peak.names$V1, ':', peak.names$V2, '-', peak.names$V3)
- pmat <- readMM(file = './data/10k_pbmc/scATAC/filtered_peak_bc_matrix/matrix.mtx')
- colnames(pmat) <- barcodes
- rownames(pmat) <- peak.names
- pmat <- pmat[,colnames([email protected]$atac)]
peak的个数还是相当多的,为了简化我们的运算量,我们需要从中挑选出不同的cluster之间差异表达的peak。
- > dim(pmat)
- [1] 94534 9642
虽然liger的官方教程说可以直接把pmat赋值给ATAC的表达矩阵,然后只用ATAC做差异表达分析即可,但我跑的时候又是各种奇怪的报错,所以变通一下,我们用seurat来构建ATAC的表达矩阵然后来做差异表达分析。
- #cell-type-specific accessibility
- #create seurat for pmat
- pmat_seurat <- CreateSeuratObject(counts = pmat,project = 'pmat')
- pmat_seurat <- NormalizeData(pmat_seurat, normalization.method = "LogNormalize", scale.factor = 10000)
- pmat_seurat <- ScaleData(pmat_seurat, features = rownames(pmat_seurat))
- cell_type <- [email protected]
- cell_type <- cell_type[colnames(pmat_seurat)]
- pmat_seurat$cell_type <- cell_type
- table(pmat_seurat$cell_type)
- Idents(pmat_seurat) <- 'cell_type'
- #find the DEPeak across the cluster
- pmat_marker <- FindAllMarkers(pmat_seurat, only.pos = TRUE, logfc.threshold = 0.25)
- pmat_marker <- pmat_marker[pmat_marker$p_val_adj < 0.05,]
- peaks.sel <- unique(pmat_marker$gene)
peaks.sel就是我们挑选出的在cluster之间差异表达的peak。我们用这组peak来过滤pmat(用于预测RNA-seq中的peak)以及pmat_seurat(用于画图)。
- #filter the pmat seurat object and scale the express
- pmat_seurat <- pmat_seurat[peaks.sel,]
- pmat_seurat <- NormalizeData(pmat_seurat, normalization.method = "LogNormalize", scale.factor = 10000)
- pmat_seurat <- ScaleData(pmat_seurat, features = rownames(pmat_seurat),do.center = FALSE)
- #filter the pmat object to impute the RNA-seq cell's ATAC peaks
- pmat <- pmat[peaks.sel,]
- int.PBMC.ds <- int.PBMC
- [email protected][['atac']] <- pmat
- int.PBMC.ds <- normalize(int.PBMC.ds)
使用ATAC-seq数据预测RNA-seq数据中的peak。
- #predict the accessibility profile for scRNA-seq data
- int.PBMC.ds <- imputeKNN(int.PBMC.ds, reference = 'atac',queries = 'rna',knn_k = 20,norm = TRUE,scale = FALSE)
预测后的数据就覆盖了原来的RNA-seq的表达谱。
- > [email protected]$rna[1:3,1:3]
- 3 x 3 sparse Matrix of class "dgCMatrix"
- rna_AAACCCAAGCGCCCAT rna_AAACCCAAGGTTCCGC
- chr6:111086981-111088830 . 0.0001584052
- chr17:82126355-82127872 0.0007113352 .
- chr8:19496551-19497709 . .
- rna_AAACCCACAGACAAGC
- chr6:111086981-111088830 4.491621e-05
- chr17:82126355-82127872 2.747334e-04
- chr8:19496551-19497709 1.823305e-04
现在我们就得到了单细胞RNA-seq数据集中细胞的转录组和(预测的)peak的特征,我们就可以去寻找基因表达与开放性元件的调控关系。想法也非常简单,我们可以计算一个基因的表达量与其上下游500kb范围内的peak强度的相关性。如果你希望扩大或者缩小这个上下游的范围,你可以用我修改过后的函数,peak_range以一个碱基为单位。
- MyLinkGenesAndPeaks <- function(gene_counts, peak_counts, genes.list = NULL, dist = "spearman",
- alpha = 0.05, path_to_coords, peak_range) {
- ## check dependency
- if (!requireNamespace("GenomicRanges", quietly = TRUE)) {
- stop("Package "GenomicRanges" needed for this function to work. Please install it by command:n",
- "BiocManager::install('GenomicRanges')",
- call. = FALSE
- )
- }
- if (!requireNamespace("IRanges", quietly = TRUE)) {
- stop("Package "IRanges" needed for this function to work. Please install it by command:n",
- "BiocManager::install('IRanges')",
- call. = FALSE
- )
- }
- ### make Granges object for peaks
- peak.names <- strsplit(rownames(peak_counts), "[:-]")
- chrs <- Reduce(append, lapply(peak.names, function(peak) {
- peak[1]
- }))
- chrs.start <- Reduce(append, lapply(peak.names, function(peak) {
- peak[2]
- }))
- chrs.end <- Reduce(append, lapply(peak.names, function(peak) {
- peak[3]
- }))
- peaks.pos <- GenomicRanges::GRanges(
- seqnames = chrs,
- ranges = IRanges::IRanges(as.numeric(chrs.start), end = as.numeric(chrs.end))
- )
- ### make Granges object for genes
- gene.names <- read.csv2(path_to_coords, sep = "t", header = FALSE, stringsAsFactors = F)
- gene.names <- gene.names[complete.cases(gene.names), ]
- genes.coords <- GenomicRanges::GRanges(
- seqnames = gene.names$V1,
- ranges = IRanges::IRanges(as.numeric(gene.names$V2), end = as.numeric(gene.names$V3))
- )
- names(genes.coords) <- gene.names$V4
- ### construct regnet
- gene_counts <- t(gene_counts) # cell x genes
- peak_counts <- t(peak_counts) # cell x genes
- # find overlap peaks for each gene
- if (missing(genes.list)) {
- genes.list <- colnames(gene_counts)
- }
- missing_genes <- !genes.list %in% names(genes.coords)
- if (sum(missing_genes)!=0){
- print(
- paste0(
- "Removing ", sum(missing_genes),
- " genes not found in given gene coordinates..."
- )
- )
- }
- genes.list <- genes.list[!missing_genes]
- genes.coords <- genes.coords[genes.list]
- print("Calculating correlation for gene-peak pairs...")
- each.len <- 0
- # assign('each.len', 0, envir = globalenv())
- elements <- lapply(seq(length(genes.list)), function(pos) {
- gene.use <- genes.list[pos]
- # re-scale the window for each gene
- gene.loci <- GenomicRanges::trim(suppressWarnings(GenomicRanges::promoters(GenomicRanges::resize(
- genes.coords[gene.use],
- width = 1, fix = "start"
- ),
- upstream = peak_range, downstream = peak_range
- )))
- peaks.use <- S4Vectors::queryHits(GenomicRanges::findOverlaps(peaks.pos, gene.loci))
- if ((x <- length(peaks.use)) == 0L) { # if no peaks in window, skip this iteration
- return(list(NULL, as.numeric(each.len), NULL))
- }
- ### compute correlation and p-adj for genes and peaks ###
- res <- suppressWarnings(psych::corr.test(
- x = gene_counts[, gene.use], y = as.matrix(peak_counts[, peaks.use]),
- method = dist, adjust = "holm", ci = FALSE, use = "complete"
- ))
- pick <- res[["p"]] < alpha # filter by p-value
- pick[is.na(pick)] <- FALSE
- if (sum(pick) == 0) { # if no peaks are important, skip this iteration
- return(list(NULL, as.numeric(each.len), NULL))
- }
- else {
- res.corr <- as.numeric(res[["r"]][pick])
- peaks.use <- peaks.use[pick]
- }
- # each.len <<- each.len length(peaks.use)
- assign('each.len', each.len length(peaks.use), envir = parent.frame(2))
- return(list(as.numeric(peaks.use), as.numeric(each.len), res.corr))
- })
- i_index <- Reduce(append, lapply(elements, function(ele) {
- ele[[1]]
- }))
- p_index <- c(0, Reduce(append, lapply(elements, function(ele) {
- ele[[2]]
- })))
- value_list <- Reduce(append, lapply(elements, function(ele) {
- ele[[3]]
- }))
- # make final sparse matrix
- regnet <- sparseMatrix(
- i = i_index, p = p_index, x = value_list,
- dims = c(ncol(peak_counts), length(genes.list)),
- dimnames = list(colnames(peak_counts), genes.list)
- )
- return(regnet)
- }
这个相关性的计算还挺漫长的,可以先去吃个饭2333。
- #find peaks and genes strongly corrected
- gmat = [email protected][['rna']]
- pseudo_pmat = [email protected][['rna']]
- regnet = MyLinkGenesAndPeaks(gene_counts = gmat, peak_counts = pseudo_pmat, dist = 'spearman', alpha = 0.05, path_to_coords = './data/10k_pbmc/annotation/genes.bed',peak_range = 500000)
对于我们关注的基因TCL1A,我们可以来看下有没有与其表达量高相关的peak。
- > which(regnet[,'TCL1A'] > 0.6)
- chr14:95456861-95457458
- 963
- > saveRDS(regnet,file = './res/fig_201112/regnet.rds')
我们成功找到了一个高相关的peak,相关性系数为0.73。为了进一步验证我们方法的可靠性,我们可以画出原始的peak的强度和RNA-seq的表达量。
- #convert to seurat and plot
- tsne.obj <- CreateDimReducObject(embeddings = [email protected][colnames(pmat_seurat),], key = "UMAP_",global = TRUE)
- pmat_seurat[['tsne']] <- tsne.obj
- TCL1A <- plotGene(int.PBMC, "TCL1A", axis.labels = c('UMAP_1', 'UMAP_2'), return.plots = TRUE)
- peak1 <- FeaturePlot(pmat_seurat,features = 'chr14:95456861-95457458',reduction = 'tsne')
- pdf(file = './res/fig_201112/high_related_peak_gene_dimplot.pdf',width = 9,height = 9)
- peak1[[1]] / TCL1A[[2]]
- dev.off()
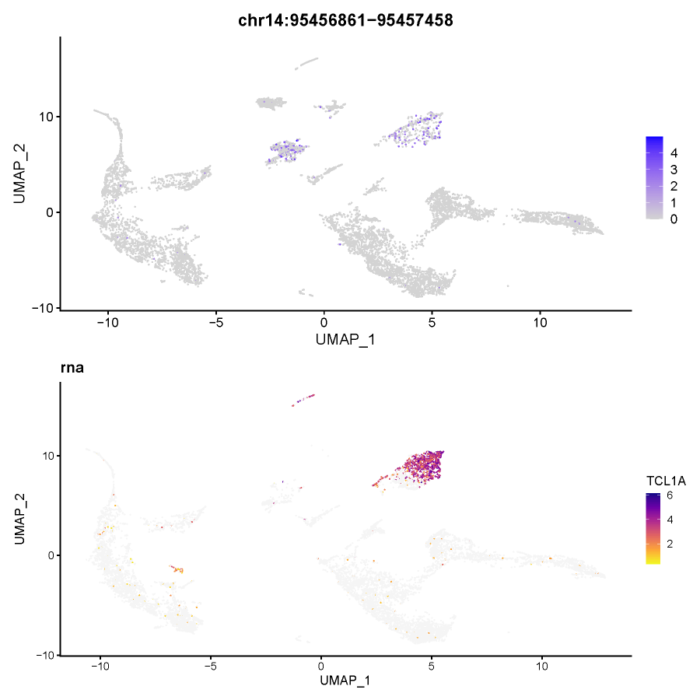
确实在B细胞群中,RNA的表达和peak的强度呈现明显的正相关,同时我们也注意到另一群细胞中peak的强度很高,但RNA的表达量却几乎没有,这其中的基因表达调控机制或许也值得我们后续的研究和探讨。
完整代码
- ############################################
- ## Project: Liger-learning
- ## Script Purpose: joint analysis of scRNA-seq and chromatin accessibility
- ## Data: 2020.10.24
- ## Author: Yiming Sun
- ############################################
- #sleep
- ii <- 1
- while(1){
- cat(paste("round",ii),sep = "n")
- ii <- ii 1
- Sys.sleep(30)
- }
- # general setting
- setwd('/data/User/sunym/project/liger_learning/')
- # library
- library(Seurat)
- library(ggplot2)
- library(dplyr)
- library(scibet)
- library(Matrix)
- library(tidyverse)
- library(cowplot)
- library(viridis)
- library(pheatmap)
- library(ComplexHeatmap)
- library(networkD3)
- library(liger)
- library(parallel)
- #my function
- MySuggestLambda <- function(object,lambda,k,knn_k = 20,thresh = 1e-04,max.iters = 100,k2 = 500,ref_dataset = NULL,resolution = 1,parl_num){
- res <- matrix(ncol = 2,nrow = length(lambda))
- colnames(res) <- c('lambda','alignment')
- res[,'lambda'] <- lambda
- cl <- makeCluster(parl_num)
- clusterExport(cl,c('object','k','knn_k','thresh','max.iters','k2','ref_dataset','resolution'),envir = environment())
- clusterExport(cl,c('optimizeALS','quantileAlignSNF','calcAlignment'),envir = .GlobalEnv)
- alignment_score <- parLapply(cl = cl,lambda,function(x){
- object <- optimizeALS(object,k = k,lambda = x,thresh = thresh,max.iters = max.iters)
- object <- quantileAlignSNF(object,knn_k = knn_k,k2 = k2,ref_dataset = ref_dataset,resolution = resolution)
- return(calcAlignment(object))
- })
- stopCluster(cl)
- res[,'alignment'] <- (alignment_score %>% unlist())
- res <- as.data.frame(res)
- res[,1] <- as.numeric(as.character(res[,1]))
- res[,2] <- as.numeric(as.character(res[,2]))
- p <- ggplot(res,aes(x = lambda,y = alignment)) geom_point() geom_line()
- return(list(res,p))
- }
- MyLinkGenesAndPeaks <- function(gene_counts, peak_counts, genes.list = NULL, dist = "spearman",
- alpha = 0.05, path_to_coords, peak_range) {
- ## check dependency
- if (!requireNamespace("GenomicRanges", quietly = TRUE)) {
- stop("Package "GenomicRanges" needed for this function to work. Please install it by command:n",
- "BiocManager::install('GenomicRanges')",
- call. = FALSE
- )
- }
- if (!requireNamespace("IRanges", quietly = TRUE)) {
- stop("Package "IRanges" needed for this function to work. Please install it by command:n",
- "BiocManager::install('IRanges')",
- call. = FALSE
- )
- }
- ### make Granges object for peaks
- peak.names <- strsplit(rownames(peak_counts), "[:-]")
- chrs <- Reduce(append, lapply(peak.names, function(peak) {
- peak[1]
- }))
- chrs.start <- Reduce(append, lapply(peak.names, function(peak) {
- peak[2]
- }))
- chrs.end <- Reduce(append, lapply(peak.names, function(peak) {
- peak[3]
- }))
- peaks.pos <- GenomicRanges::GRanges(
- seqnames = chrs,
- ranges = IRanges::IRanges(as.numeric(chrs.start), end = as.numeric(chrs.end))
- )
- ### make Granges object for genes
- gene.names <- read.csv2(path_to_coords, sep = "t", header = FALSE, stringsAsFactors = F)
- gene.names <- gene.names[complete.cases(gene.names), ]
- genes.coords <- GenomicRanges::GRanges(
- seqnames = gene.names$V1,
- ranges = IRanges::IRanges(as.numeric(gene.names$V2), end = as.numeric(gene.names$V3))
- )
- names(genes.coords) <- gene.names$V4
- ### construct regnet
- gene_counts <- t(gene_counts) # cell x genes
- peak_counts <- t(peak_counts) # cell x genes
- # find overlap peaks for each gene
- if (missing(genes.list)) {
- genes.list <- colnames(gene_counts)
- }
- missing_genes <- !genes.list %in% names(genes.coords)
- if (sum(missing_genes)!=0){
- print(
- paste0(
- "Removing ", sum(missing_genes),
- " genes not found in given gene coordinates..."
- )
- )
- }
- genes.list <- genes.list[!missing_genes]
- genes.coords <- genes.coords[genes.list]
- print("Calculating correlation for gene-peak pairs...")
- each.len <- 0
- # assign('each.len', 0, envir = globalenv())
- elements <- lapply(seq(length(genes.list)), function(pos) {
- gene.use <- genes.list[pos]
- # re-scale the window for each gene
- gene.loci <- GenomicRanges::trim(suppressWarnings(GenomicRanges::promoters(GenomicRanges::resize(
- genes.coords[gene.use],
- width = 1, fix = "start"
- ),
- upstream = peak_range, downstream = peak_range
- )))
- peaks.use <- S4Vectors::queryHits(GenomicRanges::findOverlaps(peaks.pos, gene.loci))
- if ((x <- length(peaks.use)) == 0L) { # if no peaks in window, skip this iteration
- return(list(NULL, as.numeric(each.len), NULL))
- }
- ### compute correlation and p-adj for genes and peaks ###
- res <- suppressWarnings(psych::corr.test(
- x = gene_counts[, gene.use], y = as.matrix(peak_counts[, peaks.use]),
- method = dist, adjust = "holm", ci = FALSE, use = "complete"
- ))
- pick <- res[["p"]] < alpha # filter by p-value
- pick[is.na(pick)] <- FALSE
- if (sum(pick) == 0) { # if no peaks are important, skip this iteration
- return(list(NULL, as.numeric(each.len), NULL))
- }
- else {
- res.corr <- as.numeric(res[["r"]][pick])
- peaks.use <- peaks.use[pick]
- }
- # each.len <<- each.len length(peaks.use)
- assign('each.len', each.len length(peaks.use), envir = parent.frame(2))
- return(list(as.numeric(peaks.use), as.numeric(each.len), res.corr))
- })
- i_index <- Reduce(append, lapply(elements, function(ele) {
- ele[[1]]
- }))
- p_index <- c(0, Reduce(append, lapply(elements, function(ele) {
- ele[[2]]
- })))
- value_list <- Reduce(append, lapply(elements, function(ele) {
- ele[[3]]
- }))
- # make final sparse matrix
- regnet <- sparseMatrix(
- i = i_index, p = p_index, x = value_list,
- dims = c(ncol(peak_counts), length(genes.list)),
- dimnames = list(colnames(peak_counts), genes.list)
- )
- return(regnet)
- }
- # 1.joint analysis of scRNA-seq and chromatin accessibility
- # pre-prapare scTATA data
- #sort by chromosome start position and end position for fragment, gene and promoter
- system('gunzip ./data/10k_pbmc/scATAC/fragments.tsv.gz')
- system('tail -n 6 ./data/10k_pbmc/annotation/gencode.v28.annotation.gtf > ./data/10k_pbmc/annotation/genes_split.gtf')
- genes <- read.table('./data/10k_pbmc/annotation/genes_split.gtf',sep = 't')
- genes <- genes[genes[,3] == 'gene',]
- gene_name <- unlist(strsplit(as.character(genes[,9]),split = '; ',fixed = TRUE))
- gene_name <- gene_name[grepl(pattern = 'gene_name',gene_name)]
- gene_name <- sub(pattern = 'gene_name ',replacement = '',gene_name)
- genes[,9] <- gene_name
- genes <- genes[,c(1,4,5,9,6,7)]
- promoter <- as.data.frame(matrix(nrow = dim(genes)[1],ncol = dim(genes)[2]))
- promoter[,c(1,4,5,6)] <- genes[,c(1,4,5,6)]
- for (i in 1:dim(promoter)[1]) {
- if(promoter[i,6] == ' '){
- if ((as.numeric(genes[i,2])-2000) >= 0){
- promoter[i,2] <- as.numeric(genes[i,2])-2000
- }else{
- promoter[i,2] <- 0
- }
- promoter[i,3] <- as.numeric(genes[i,2]) 2000
- } else if(promoter[i,6] == '-'){
- if ((as.numeric(genes[i,3])-2000) >= 0){
- promoter[i,2] <- as.numeric(genes[i,3])-2000
- }else{
- promoter[i,2] <- 0
- }
- promoter[i,3] <- as.numeric(genes[i,3]) 2000
- }
- }
- write.table(genes,file = './data/10k_pbmc/annotation/genes.bed',sep = 't',col.names = FALSE,row.names = FALSE,quote = FALSE)
- write.table(promoter,file = './data/10k_pbmc/annotation/promoter.bed',sep = 't',col.names = FALSE,row.names = FALSE,quote = FALSE)
- system('/data/User/sunym/software/BEDOPS/bin/sort-bed --max-mem 50G ./data/10k_pbmc/scATAC/fragments.tsv > ./data/10k_pbmc/scATAC/atac_fragments.sort.bed')
- system('/data/User/sunym/software/BEDOPS/bin/sort-bed --max-mem 50G ./data/10k_pbmc/annotation/genes.bed > ./data/10k_pbmc/annotation/genes.sort.bed')
- system('/data/User/sunym/software/BEDOPS/bin/sort-bed --max-mem 50G ./data/10k_pbmc/annotation/promoter.bed > ./data/10k_pbmc/annotation/promoters.sort.bed')
- #Use bedmap command to calculate overlapping elements between indexes and fragment output files
- system('/data/User/sunym/software/BEDOPS/bin/bedmap --ec --delim "t" --echo --echo-map-id ./data/10k_pbmc/annotation/promoters.sort.bed ./data/10k_pbmc/scATAC/atac_fragments.sort.bed > ./data/10k_pbmc/scATAC/atac_promoters_bc.bed')
- system('/data/User/sunym/software/BEDOPS/bin/bedmap --ec --delim "t" --echo --echo-map-id ./data/10k_pbmc/annotation/genes.sort.bed ./data/10k_pbmc/scATAC/atac_fragments.sort.bed > ./data/10k_pbmc/scATAC/atac_genes_bc.bed')
- # load data
- #load scATAC peak filted barcode data
- barcodes <- read.table(file = './data/10k_pbmc/scATAC/filtered_peak_bc_matrix/barcodes.tsv',sep = 't',as.is = TRUE,header = FALSE)$V1
- #import the bedmap outputs into the R
- genes.bc <- read.table(file = "./data/10k_pbmc/scATAC/atac_genes_bc.bed", sep = "t", as.is = c(4,7), header = FALSE)
- promoters.bc <- read.table(file = "./data/10k_pbmc/scATAC/atac_promoters_bc.bed", sep = "t", as.is = c(4,7), header = FALSE)
- #filter the barcode with bad quality
- bc <- genes.bc[,7]
- bc_split <- strsplit(bc,";")
- bc_split_vec <- unlist(bc_split)
- bc_counts <- table(bc_split_vec)
- bc_filt <- names(bc_counts)[bc_counts > 1500]
- barcodes <- intersect(bc_filt,barcodes)
- #use LIGER's makeFeatureMatrix function to calculate accessibility counts for gene body and promoter
- gene.counts <- makeFeatureMatrix(genes.bc, barcodes)
- promoter.counts <- makeFeatureMatrix(promoters.bc, barcodes)
- gene.counts <- gene.counts[order(rownames(gene.counts)),]
- promoter.counts <- promoter.counts[order(rownames(promoter.counts)),]
- PBMCs <- gene.counts promoter.counts
- colnames(PBMCs)=paste0("PBMC_",colnames(PBMCs))
- #load scRNA-seq data
- PBMC.rna <- read10X(sample.dirs = './data/10k_pbmc/scRNA/filtered_feature_bc_matrix/',sample.names = 'rna')
- #create a LIGER object
- PBMC.data <- list(atac = PBMCs, rna = PBMC.rna)
- int.PBMC <- createLiger(PBMC.data)
- remove(list = ls()[ls() != 'int.PBMC'])
- gc()
- #normalize select gene and scale
- int.PBMC <- normalize(int.PBMC)
- int.PBMC <- selectGenes(int.PBMC, datasets.use = 2)
- int.PBMC <- scaleNotCenter(int.PBMC)
- #integrate NMF
- #suggest lambda
- suggestLambda(int.PBMC,lambda.test = seq(1,20,1),k = 25,num.cores = 10)
- #suggest k
- suggestK(object = int.PBMC,k.test = seq(10,30,1),lambda = 10,num.cores = 10,plot.log2 = FALSE)
- #lambda = 10,k = 25
- int.PBMC <- optimizeALS(int.PBMC,k = 25,lambda = 10,max.iters = 30,thresh = 1e-06)
- #Quantile Normalization and Joint Clustering
- int.PBMC <- quantile_norm(int.PBMC,knn_k = 20,quantiles = 50,min_cells = 20,do.center = FALSE,
- max_sample = 1000,refine.knn = TRUE,eps = 0.9)
- int.PBMC <- louvainCluster(int.PBMC, resolution = 0.4)
- table([email protected])
- #Visualization and Downstream Analysis
- int.PBMC <- runUMAP(int.PBMC)
- all.plots <- plotByDatasetAndCluster(int.PBMC, axis.labels = c('UMAP 1', 'UMAP 2'),return.plots = TRUE)
- head(all.plots[[1]]$data)
- pdf(file = './res/fig_201112/plot_by_dataset_and_cluster.pdf',width = 8,height = 4)
- all.plots[[1]] all.plots[[2]]
- dev.off()
- #identify marker genes for each cluster
- int.PBMC.wilcoxon <- runWilcoxon(int.PBMC, data.use = 'all', compare.method = 'clusters')
- #filter the enriched gene yourself
- int.PBMC.wilcoxon <- int.PBMC.wilcoxon[int.PBMC.wilcoxon$padj < 0.05,]
- int.PBMC.wilcoxon <- int.PBMC.wilcoxon[int.PBMC.wilcoxon$logFC > 3,]
- #top 20 markers of cluster 3 and 4
- wilcoxon.cluster_3 <- int.PBMC.wilcoxon[int.PBMC.wilcoxon$group == 3, ]
- wilcoxon.cluster_3 <- wilcoxon.cluster_3[order(wilcoxon.cluster_3$padj), ]
- markers.cluster_3 <- wilcoxon.cluster_3[1:20, ]
- wilcoxon.cluster_4 <- int.PBMC.wilcoxon[int.PBMC.wilcoxon$group == 4, ]
- wilcoxon.cluster_4 <- wilcoxon.cluster_4[order(wilcoxon.cluster_4$padj), ]
- markers.cluster_4 <- wilcoxon.cluster_4[1:20, ]
- #feature plot on umap
- # CD8A for cluster 2 and CD79A for cluster 4
- CD8A <- plotGene(int.PBMC, "CD8A", axis.labels = c('UMAP 1', 'UMAP 2'), return.plots = TRUE)
- CD79A <- plotGene(int.PBMC, "CD79A", axis.labels = c('UMAP 1', 'UMAP 2'), return.plots = TRUE)
- pdf(file = './res/fig_201112/marker_for_cluster_3_and_4.pdf',width = 9,height = 6)
- plot_grid(CD8A[[2]],CD79A[[2]],CD8A[[1]],CD79A[[1]], ncol=2)
- dev.off()
- #gene loading plot
- # gene loading plot of factor12
- all.plots <- plotGeneLoadings(int.PBMC, return.plots = TRUE,do.spec.plot = FALSE)
- pdf(file = './res/fig_201112/gene_loading_factor_18.pdf')
- all.plots[[18]]
- dev.off()
- # feature plot
- TCL1A <- plotGene(int.PBMC, "TCL1A", axis.labels = c('UMAP 1', 'UMAP 2'), return.plots = TRUE)
- RPS9 <- plotGene(int.PBMC, "RPS9", axis.labels = c('UMAP 1', 'UMAP 2'), return.plots = TRUE)
- pdf(file = './res/fig_201112/RPS9_TCL1A_plot_for_RNA_and_ATAC.pdf',width = 9,height = 6)
- plot_grid(RPS9[[2]],TCL1A[[2]],RPS9[[1]],TCL1A[[1]], ncol=2)
- dev.off()
- # Pseudo single multi-omic data (scRNA-seq and scATAC-seq)
- #load scATAC peak data
- barcodes <- read.table(file = './data/10k_pbmc/scATAC/filtered_peak_bc_matrix/barcodes.tsv',sep = 't',as.is = TRUE,header = FALSE)$V1
- barcodes <- paste('PBMC',barcodes,sep = '_')
- peak.names <- read.table(file = './data/10k_pbmc/scATAC/filtered_peak_bc_matrix/peaks.bed', sep = 't', header = FALSE)
- peak.names <- paste0(peak.names$V1, ':', peak.names$V2, '-', peak.names$V3)
- pmat <- readMM(file = './data/10k_pbmc/scATAC/filtered_peak_bc_matrix/matrix.mtx')
- colnames(pmat) <- barcodes
- rownames(pmat) <- peak.names
- pmat <- pmat[,colnames([email protected]$atac)]
- #cell-type-specific accessibility
- #create seurat for pmat
- pmat_seurat <- CreateSeuratObject(counts = pmat,project = 'pmat')
- pmat_seurat <- NormalizeData(pmat_seurat, normalization.method = "LogNormalize", scale.factor = 10000)
- pmat_seurat <- ScaleData(pmat_seurat, features = rownames(pmat_seurat))
- cell_type <- [email protected]
- cell_type <- cell_type[colnames(pmat_seurat)]
- pmat_seurat$cell_type <- cell_type
- table(pmat_seurat$cell_type)
- Idents(pmat_seurat) <- 'cell_type'
- #find the DEPeak across the cluster
- pmat_marker <- FindAllMarkers(pmat_seurat, only.pos = TRUE, logfc.threshold = 0.25)
- pmat_marker <- pmat_marker[pmat_marker$p_val_adj < 0.05,]
- peaks.sel <- unique(pmat_marker$gene)
- #filter the pmat seurat object and scale the express
- pmat_seurat <- pmat_seurat[peaks.sel,]
- pmat_seurat <- NormalizeData(pmat_seurat, normalization.method = "LogNormalize", scale.factor = 10000)
- pmat_seurat <- ScaleData(pmat_seurat, features = rownames(pmat_seurat),do.center = FALSE)
- #filter the pmat object to impute the RNA-seq cell's ATAC peaks
- pmat <- pmat[peaks.sel,]
- int.PBMC.ds <- int.PBMC
- [email protected][['atac']] <- pmat
- int.PBMC.ds <- normalize(int.PBMC.ds)
- #predict the accessibility profile for scRNA-seq data
- int.PBMC.ds <- imputeKNN(int.PBMC.ds, reference = 'atac',queries = 'rna',knn_k = 20,norm = TRUE,scale = FALSE)
- #find peaks and genes strongly corrected
- gmat = [email protected][['rna']]
- pseudo_pmat = [email protected][['rna']]
- regnet = MyLinkGenesAndPeaks(gene_counts = gmat, peak_counts = pseudo_pmat, dist = 'spearman', alpha = 0.05, path_to_coords = './data/10k_pbmc/annotation/genes.bed',peak_range = 500000)
- which(regnet[,'TCL1A'] > 0.6)
- saveRDS(regnet,file = './res/fig_201112/regnet.rds')
- #convert to seurat and plot
- tsne.obj <- CreateDimReducObject(embeddings = [email protected][colnames(pmat_seurat),], key = "UMAP_",global = TRUE)
- pmat_seurat[['tsne']] <- tsne.obj
- TCL1A <- plotGene(int.PBMC, "TCL1A", axis.labels = c('UMAP_1', 'UMAP_2'), return.plots = TRUE)
- peak1 <- FeaturePlot(pmat_seurat,features = 'chr14:95456861-95457458',reduction = 'tsne')
- pdf(file = './res/fig_201112/high_related_peak_gene_dimplot.pdf',width = 9,height = 9)
- peak1[[1]] / TCL1A[[2]]
- dev.off()
写在最后
在写这篇博客的时候遇到了不少的困难,liger作者给出的教程中基因组注释文件似乎不是特别匹配,导致我最终重复不出他们的结果。所以我决定从头开始,自己收集数据和reference,从而构建出一套完整的流程。可能我对外周血单核细胞的认知也比较浅薄,举例时所选择的基因和peak也不一定恰当,如果读者发现有什么错误之处,希望能在评论区指出,大家一起进步。