

chromVAR是一个用于分析稀疏染色质开放的R包。chromVAR的输入文件包括,ATAC-seq处理后的fragments文件(过滤重复和低质量数据), DNAse-seq实验结果,以及基因组注释(例如motif位置)
chromVAR先根据所有细胞或者样本的平均情况来计算期望开放性, 然后用它来计算每个注释,每个细胞或样本的偏差,最后对开放进行纠正。
安装加载
安装R包
- BiocManager::install("GreenleafLab/chromVAR")
- BiocManager::install("motifmatchr")
- BiocManager::install(SummarizedExperiment)
- BiocManager::install(BiocParallel)
- BiocManager::install("JASPAR2018") # JASPAR 2018数据库
chromVAR的运行需要先加载下面这些R包
- library(chromVAR)
- library(motifmatchr)
- library(Matrix)
- library(SummarizedExperiment)
- library(BiocParallel)
- set.seed(2019)
chromVAR能够使用多核进行并行运算,调用方法如下
- # 全部用户
- register(MulticoreParam(8, progressbar = TRUE))
- # Windows用户,MulticoreParam不好用就用SnowParam
- register(SnowParam(workers = 1, type = "SOCK"))
注意: 不运行的话,后续代码可能会报错
读取输入
chromVAR接受的输入是落入开放区域的read数统计表。有许多软件可以做到,chromVAR也提供了相应的方法。
首先要提供一个peak文件,文档建议这个peak文件存放的peak为等宽非重叠,建议宽度在250-500 bp之间。peak文件可以利用已有的bulk ATAC-seq或DNAse-seq数据来获取。对于来自于多个样本的peak,需要先用filterPeaks函数保证peak之间不重合。
使用getPeaks读取peak
- peakfile <- system.file("extdata/test_bed.txt", package = "chromVAR")
- peaks <- getPeaks(peakfile, sort_peaks = TRUE)
MACS2分析结果里会提供narrowpeak格式文件,chromVAR提供了readNarrowpeaks函数进行读取。
随后用getCounts函数基于BAM文件和加载的peak获取count
- bamfile <- system.file("extdata/test_RG.bam", package = "chromVAR")
- fragment_counts <- getCounts(bamfile,
- peaks,
- paired = TRUE,
- by_rg = TRUE,
- format = "bam",
- colData = DataFrame(celltype = "GM"))
bamfile可以有多个bam文件路径,bam文件的类型和colData对应。此外by_rg定义是否要根据BAM文件中的RG对输入分组。
实际演示的时候,我们官方提供的示例数据
data(example_counts, package = "chromVAR")
因为这是一个*SummarizedExperiment 对象,因此我们就能用assay,colData和rowData 了解这个数据集
主体是一个存放count的dgCMatrix
- assay(example_counts)[1:5,1:2]
- 5 x 2 sparse Matrix of class "dgCMatrix"
- singles-GM12878-140905-1 singles-GM12878-140905-2
- [1,] . 1
- [2,] . .
- [3,] . .
- [4,] . .
- [5,] . .
行是特征(feature), 这里就是peak
- head(rowData(example_counts), n=2)
- DataFrame with 2 rows and 3 columns
- score qval name
- 1 259 25.99629 GM_peak_6
- 2 82 8.21494 H1_peak_
列表示样本,
- head(colData(example_counts), n=2)
- DataFrame with 2 rows and 2 columns
- Cell_Type depth
- singles-GM12878-140905-1 GM 9220
- singles-GM12878-140905-2 GM 9401
前期准备
分析peak中的GC含量
GC含量信息用于确定哪些peak可能是背景, addGCBias返回更新后的SummarizedExperiment 。其中genome支持BSgenome, FaFile和DNAStringSet对象。
- library(BSgenome.Hsapiens.UCSC.hg19)
- example_counts <- addGCBias(example_counts,
- genome = BSgenome.Hsapiens.UCSC.hg19)
- head(rowData(example_counts))
过滤输入(单细胞)
如果是处理单细胞数据,建议过滤测序深度不够,或者是信噪比低不够(也就是peak中所占read比例低)的数据,主要靠两个参数,min_in_peaks和min_depth.
- counts_filtered <- filterSamples(example_counts, min_depth = 1500,
- min_in_peaks = 0.15, shiny = FALSE)
然后我们用filterSamplesPlot对过滤情况进行可视化
- filtering_plot <- filterSamplesPlot(example_counts, min_depth = 1500,
- min_in_peaks = 0.15, use_plotly = FALSE)
- filtering_plot
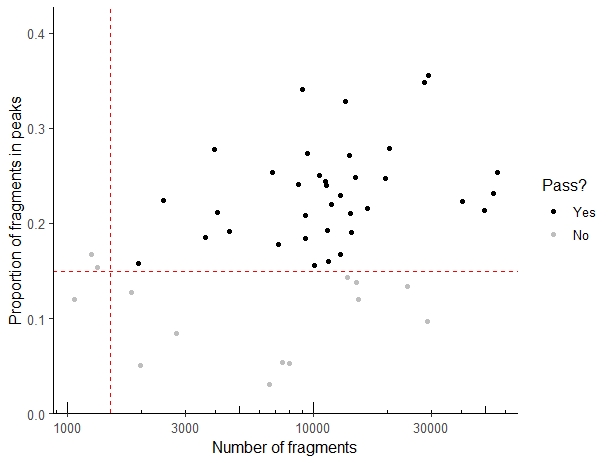
确认标准后,就可以过滤
counts_filtered <- filterPeaks(counts_filtered, non_overlapping = TRUE)
获取Motifs和分析包含motif的peak
可以用getJasparMotifs在JASPAR数据库中提取motif信息,但是其中getJasparMotifs只是TFBSTools::getMatrixSet一个简单的封装而已, 默认用的是JASPAR2016, 建议通过下面的代码获取最新版本的数据。
- # BiocManager::install("JASPAR2018")
- library(JASPAR2018)
- species <- "Homo sapiens"
- collection <- "CORE"
- opts <- list()
- opts["species"] <- species
- opts["collection"] <- collection
- out <- TFBSTools::getMatrixSet(JASPAR2018, opts)
- if (!isTRUE(all.equal(TFBSTools::name(out), names(out))))
- names(out) <- paste(names(out), TFBSTools::name(out),
- sep = "_")
- motif <- out
collection参数中接受: “CORE”, “CNE”, “PHYLOFACTS”, “SPLICE”, “POLII”, “FAM”, “PBM”, “PBM_HOMEO”, “PBM_HLH” 选项。
获取Motif的另外一种方式是用chromVARmotifs, 这个R包的主要功能就是整合了一些motif, 通过devtools::install_github("GreenleafLab/chromVARmotifs")进行安装。
之后用motifmatchr中的macthMotifs去分析peak中motif包含情况。默认会返回一个SummarizedExperiment 对象,就是一个稀疏矩阵,来提示是不是匹配了motif
- library(motifmatchr)
- motif_ix <- matchMotifs(motifs, counts_filtered,
- genome = BSgenome.Hsapiens.UCSC.hg19)
关键参数是p.cutoff用于设置严格度,默认是0.00005其实能够返回比较合理的结果。如果需要一些额外信息,可以通过参数out来调整,例如out=positions表示返回实际的匹配位置
偏离度和变异度分析(Deviations and Variability )
上面都是准备阶段,计算deviations和Variability才是这个软件的重点。
计算偏离度
函数computeDeviations返回的SummarizedExperiment 对象, 包含两个"assays",
- 偏差纠正后的开放偏离度:
assays(dev)$deviations - 偏离度的Z-score:
assays(deviations)$z: 考虑到了当从基因组上随机抽取一些GC含量类似的片段分析时,有多大概率会得到该结果。
dev <- computeDeviations(object = counts_filtered, annotations = motif_ix)
compuateDeviations支持背景peak的输入,用于的偏移度得分进行标准化,默认会自动计算不会返回结果。你可以选择用getBackgroundPeaks分析背景,然后传递给computeDeviations
- bg <- getBackgroundPeaks(object = counts_filtered)
- dev <- computeDeviations(object = counts_filtered, annotations = motif_ix,
- background_peaks = bg)
变异度
computeVariability会返回一个数据框data.frame,里面有变异度, 该变异度的bootstrap置信区间和衡量拒绝原假设的概率(即 > 1)
- variability <- computeVariability(dev)
- plotVariability(variability, use_plotly = FALSE)
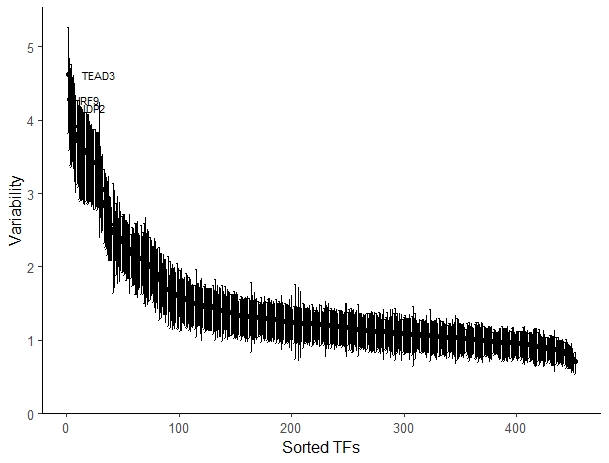
偏离度可视化
可以利用tSNE可视化偏离度
- tsne_results <- deviationsTsne(dev, threshold = 1.5, perplexity = 10)
- tsne_plots <- plotDeviationsTsne(dev, tsne_results,
- annotation_name = "TEAD3",
- sample_column = "Cell_Type",
- shiny = FALSE)
- cowplot::plot_grid(tsne_plots[[1]],tsne_plots[[1]] )
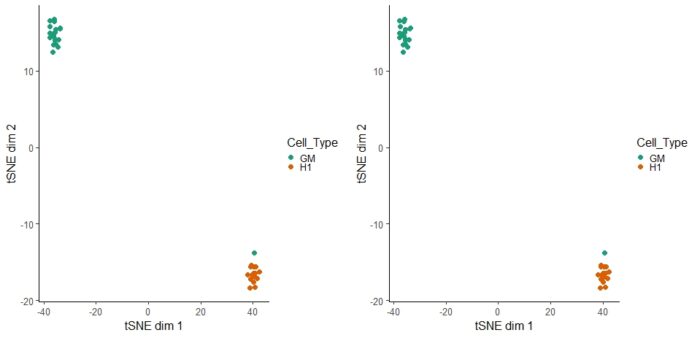
参考资料
- 数据库JASPAR是一个开放性数据库,用于存放人工审查的非冗余转录因子(TF)结合谱,数据格式为PFM(position frequency matrices ), TFFM( TF flexible models)
- chromVAR官方文档
- chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data