

什么是遗传变异
遗传变异(genetic variation)是指一个群体中个体之间DNA序列的差异。变异可发生在生殖细胞(即精子和卵子)中,也发生在体细胞(所有其他)中。只有在生殖细胞中产生的变异才能从一个个体遗传给另一个个体,从而影响种群的动态,并最终影响进化。
突变(Mutation)和重组(Recombination)是变异的主要来源。
遗传变异的类型
- 点突变Single base-pair mutation只有一个碱基发生了替换,具体包括Transition和Transversion。Transition指嘌呤(A/G)之间或嘧啶(T/C)之间的替换。Transversion指嘌呤和嘧啶间的替换。
- 插入/缺失(Indel)主要指在基因组某个位置上发生较短长度的线性片段插入或者删除的现象。强调线性的原因是,这里的插入和删除是有前后顺序的与下述的结构性变异不同。Indel长度通常在50bp以下,更多时候甚至是不超过10bp,这个长度范围内的序列变化可以通过Smith-Waterman 的局部比对算法来准确获得,并且也能够在目前短读长的测序数据中较好地检测出来。
- 结构变异通常就是指基因组上大长度的序列变化和位置关系变化。类型很多,包括长度在50bp以上的长片段序列插入或者删除(Big Indel)、串联重复(Tandem repeate)、染色体倒位(Inversion)、染色体内部或染色体之间的序列易位(Translocation)、拷贝数变异(CNV)以及形式更为复杂的嵌合性变异。1Kb与3Mb之间的序列,包括插入、缺失、拷贝数变异、倒位和易位。
变异的鉴定和分析
变异的鉴定主要分为两个步骤:比对和variants calling
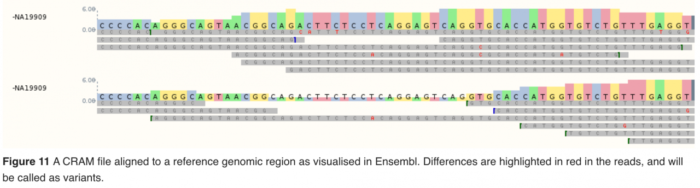
- 比对 常用的软件有:BWA,Bowtie2。将质控后的数据(fastq)比对到参考基因组上,得到比对后的reads(bam或CRAM格式)。
- variants calling常用的软件有GATK, bcftools, FreeBays等,这里的variants通常只包含SNPs和Indels,最终得到vcf格式的文件。
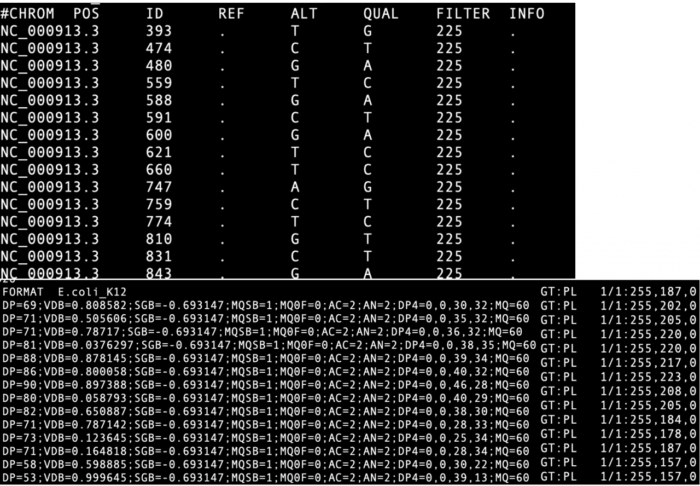
- vcf文件格式介绍vcf是Variant Call Format的简称,即是变异文件储存的标准格式。下图是E.coli 变异分析的vcf文件示例。
- CHROM: 染色体,由于E.coli是一个环形的基因组,这里没有区分染色体
- POS: 变异位点
- REF: 参考基因组位点的碱基
- ALT:个体变异位点的碱基
- QUAL:质量
- FILTER:如果使用GATK过滤后,会对通过过滤的变异位点有PASS的标签
- INFO: 这一栏包含的信息较多,其中
- DP:表示覆盖在这个位点的总reads数,也就是这个位点的测序深度
- GT: 表示genotype,通常用”/” or “|”分隔两个数字,“|”phase过也就是杂合的两个等位基因知道哪个等位基因来自哪条染色体;0代表参考基因组的碱基类型;1代表ALT碱基类型的第一个碱基(多个碱基用”,”分隔),2代表ALT第二个碱基,以此类推;比如 REF列为:A, ALT列为G,T;那么0/1基因型为AG 杂合,1/1基因型为GG纯合SNP;1/2代表GT基因型;./.表示缺失。
变异分析常用的工具:bcftools & vcftools
bcftools的部分功能介绍
- Calling Variants
- bcftools mpileup -f REF.fa sample.sorted.bam | bcftools call -vm -Oz > sample.vcf.gz
- 提取等位基因和基因型信息
- bcftools query -f '%CHROM %POS %REF %ALT [%TGT]n' query.vcf.gz -o query.extract.txt
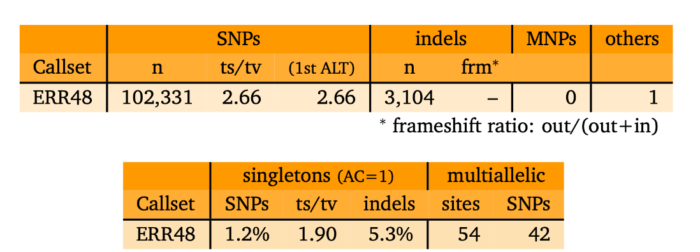
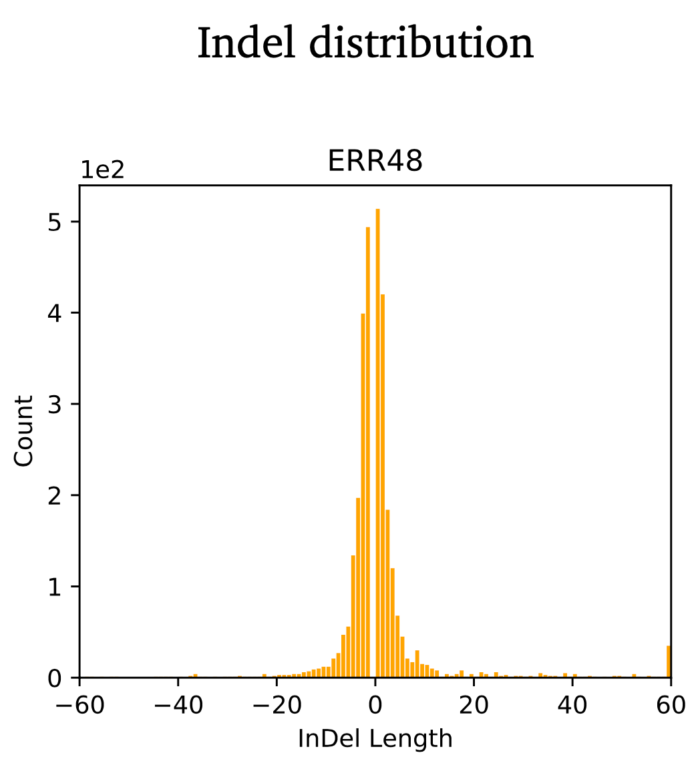
- 变异位点的基本统计分析
- bcftools stats sample.vcf > sample.vcf.stats
- plot-vcfstats sample.vcf.stats -p sample.vcf.stats.plot_output
vcftools的部分功能介绍
- vcf文件的过滤
- vcftools --vcf sample.vcf --minDP 4 --max-missing 0.2 --minQ 30 --recode --recode-INFO-all --out filter
max-missing 0.5: 过滤低于50%的基因型--minDP 4:过滤depth低于4的reads
--recode: 标志告诉程序使用过滤器写入一个新的vcf文件,--recode-INFO-all保留旧vcf文件中的所有INFO标志。--out输出的文件名称的前缀 - 拆分SNPs和Indels
- ### 保留SNPs
- vcftools --vcf filter.recode.vcf --remove-indels --recode --recode-INFO-all --out filter.snps
- ### 保留Indels
- vcftools --vcf filter.recode.vcf --keep-only-indels --recode --recode-INFO-all --out filter.indels