

ATAC-Seq分析教程系列
ATAC-Seq分析教程:ATAC-seq的背景介绍以及与ChIP-Seq的异同
ATAC-Seq分析教程:用MACS2软件call peaks
ATAC-Seq分析教程:对ATAC-Seq/ChIP-seq的质量评估(一)phantompeakqualtools
ATAC-Seq分析教程:对ATAC-Seq/ChIP-seq的质量评估(二)ChIPQC
ATAC-Seq分析教程:用ChIPseeker对peaks进行注释和可视化
ATAC-Seq分析教程:用网页版工具做功能分析和motif分析
这部分内容包括对原始测序数据质控,然后比对过滤,这是所有NGS数据处理的上游分析。
- ATAC-Seq与其他方法不同的一点是需要过滤去除线粒体(如果是植物,还需要过滤叶绿体),因为线粒体DNA是裸露的,也可以被Tn5酶识别切割。
- 另外一点需要注意的是课程中给出的是单端比对的示例代码,如果是双端测序做相应更改即可。
学习目标
- 用FastQC进行质控检测
- 用Trimmomatic进行质量过滤
- 用Bowtie2比对,并理解相关参数含义
测序reads 的质控流程示意图
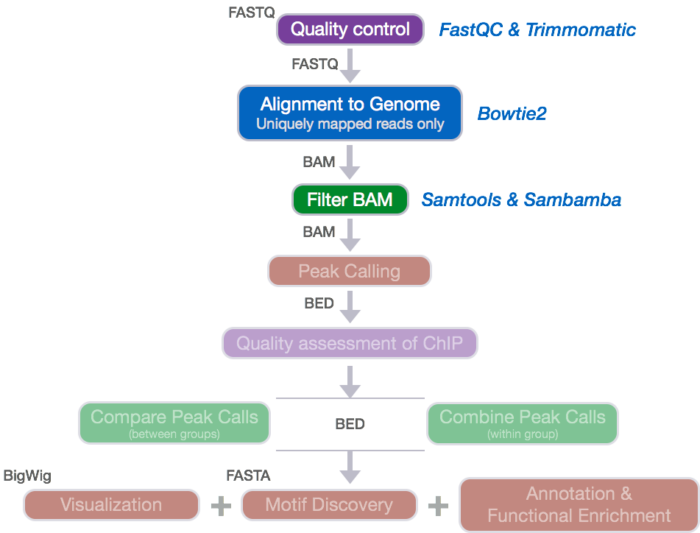
FASTQC
首先对拿到的原始测序数据(fastq或fastq.gz格式)进行质控检测,直接用fastqc软件,再加上multiqc将多个检测结果一起展示。
如:
- fastqc -o out_dir raw_data/*gz
- multiqc *fastqc.zip --ignore *.html
Trimmomatic
Trimmomatic 可以用于去除接头,过滤低质量数据。相同功能的软件还有很多,如trim_galore、cutadapt等,个人比较喜欢trim_galore可以自动识别接头类型。
- # 课程中给出的Trimmomatic 的用法(单端测序) $ java -jar /opt/Trimmomatic-0.33/trimmomatic-0.33.jar SE -threads 2 -phred33 H1hesc_Input_Rep1_chr12.fastq ../results/trimmed/H1hesc_Input_Rep1_chr12.qualtrim20.minlen36.fq LEADING:20 TRAILING:20 MINLEN:36
Trimmomatic参数含义:可以参考NGS 数据过滤之 Trimmomatic 详细说明
trim_galore使用示例
- trim_galore -q 20 --phred33 --stringency 3 --length 20 -e 0.1 --paired fq1 fq2 --gzip -o input_data_dir
- # 重新用fastqc检测进行过滤后的reads质量
- fastqc -o out_dir *fq.gz
- multiqc *fastqc.zip --ignore *.html
比对
Bowtie2是一个快速精确的比对工具,基于Burrows-Wheeler Transform 构建基因组的FM 索引,比对过程所耗内存少。Bowtie2支持局部、双端、缺口比对模式,对大于50bp的reads比对效果更好(小于50bp的reads用Bowtie1)。
创建Bowtie2索引
- bowtie2-build
- # Can find indexes for the entire genome on Orchestra using following path: /groups/shared_databases/igenome/Homo_sapiens/UCSC/hg19/Sequence/Bowtie2Index/
Bowtie2 比对
- p: 线程数
- q: reads是fastq格式
- x: index路径
- U: fastq路径
- S: 输出Sam格式文件
- ## 课程中给出的代码是单端比对
- bowtie2 -p 2 -q -x ~/ngs_course/chipseq/reference_data/chr12 -U ~/ngs_course/chipseq/results/trimmed/H1hesc_Input_Rep1_chr12.qualtrim20.minlen36.fq -S ~/ngs_course/chipseq/results/bowtie2/H1hesc_Input_Rep1_chr12_aln_unsorted.sam
NOTE: 如果fastq文件是没有经过trim的,可以用局部比对执行soft-clipping,加上参数--local
过滤reads
首先将sam文件转为bam格式,再对bam文件进行排序,接着过滤唯一比对的reads,去除线粒体reads。
转化为bam格式
使用samtools转换格式
- samtools view -h -S -b -o H1hesc_Input_Rep1_chr12_aln_unsorted.bam H1hesc_Input_Rep1_chr12_aln_unsorted.sam
对bam文件排序
对bam文件按照基因组坐标排序,可以直接使用samtools,也可以使用Sambamba。sambamba快速处理bam和sam文件。
- sambamba sort -t 2
- -o H1hesc_Input_Rep1_chr12_aln_sorted.bam
- H1hesc_Input_Rep1_chr12_aln_unsorted.bam
过滤唯一比对的reads
- sambamba view -h -t 2 -f bam
- -F "[XS] == null and not unmapped "
- H1hesc_Input_Rep1_chr12_aln_sorted.bam > H1hesc_Input_Rep1_chr12_aln.bam
去除PCR重复
PCR扩增和一些重复序列(如微卫星、着丝粒)会产生重复,干扰真实的富集信号,所以在call peaks前需要先去除重复,这里先用picard去除PCR重复。picard去除PCR重复时要加上参数REMOVE_DUPLICATES=true,否则只是标记了duplicates,并没有去除。
- java -jar picard-tools-1.119/MarkDuplicates.jar REMOVE_DUPLICATES=true I=H1hesc_Input_Rep1_chr12_aln.bam O=H1hesc_Input_Rep1_chr12_aln.dedup.bam M=H1hesc.duplicates.log
过滤线粒体reads
- samtools index H1hesc_Input_Rep1_chr12_aln.dedup.bam
- samtools idxstats H1hesc_Input_Rep1_chr12_aln.dedup.bam > H1hesc_Input_Rep1_chr12_aln.dedup.mitochondrial.stats
- samtools view -h H1hesc_Input_Rep1_chr12_aln.dedup.bam | grep -v 'chrM' | samtools view -bS -o H1hesc.final.bam
上面给出的仅是示例代码,和参考课程不一样,实际运行需要修改相应文件。
此时就得到了唯一比对且已经去除过线粒体的比对文件,可以用于接下来的peaks calling。
参考资料:
HBC的深度NGS数据分析课程:https://github.com/hbctraining/In-depth-NGS-Data-Analysis-Course/blob/master/sessionV/lessons/02_QC_and_alignment.md