

ATAC-Seq分析教程系列
ATAC-Seq分析教程:ATAC-seq的背景介绍以及与ChIP-Seq的异同
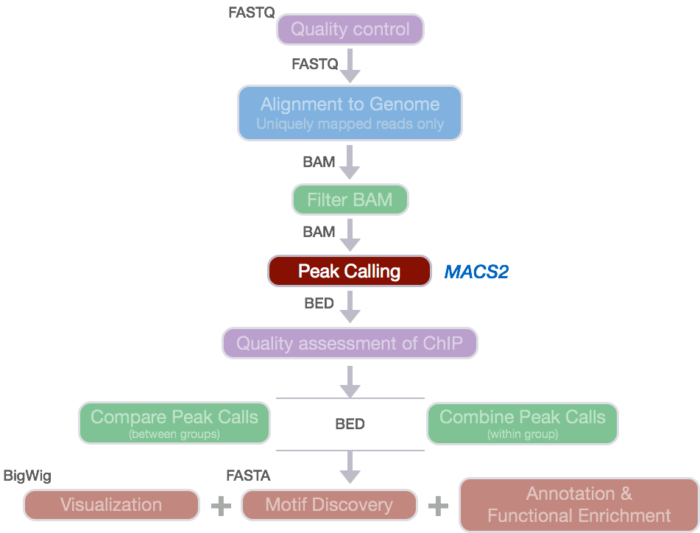
ATAC-Seq分析教程:用MACS2软件call peaks
ATAC-Seq分析教程:对ATAC-Seq/ChIP-seq的质量评估(一)phantompeakqualtools
ATAC-Seq分析教程:对ATAC-Seq/ChIP-seq的质量评估(二)ChIPQC
ATAC-Seq分析教程:用ChIPseeker对peaks进行注释和可视化
ATAC-Seq分析教程:用网页版工具做功能分析和motif分析
学习目标:
- 学会用MACS2 call peaks
- 理解MACS2 call peaks的结果
Peak Calling
Peak calling即利用计算的方法找出ChIP-seq或ATAC-seq中reads富集的基因组区域。
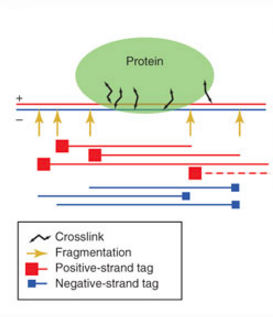
如下图所示,比对结果的文件中reads在正负链不均匀分布,但在结合位点聚集。正负链5‘末端的reads各形成一组合,通过统计学的方法评估这些组合的分布并和对照组比较,确定这些结合位点是否是显著的。
NOTE:ChIP-seq的分析方法可以鉴定两种类型的富集模式:broad domains和narrow peaks。broad domains,如组蛋白修饰在整个基因body区域的分布;narrow peak,如转录因子的结合。narrow peak相对于broad 或者分散的marks更易被检测到。也有一些混合的结合图谱,如PolII包括narrow和broad信号。
MACS2
peaks calling 有不同的方法,MACS2是最常用的call peaks工具。 MACS全称Model-based Analysis of ChIP-Seq,最初的设计是用来鉴定转录因子的结合位点,但是它也可以用于其他类型的富集方式测序。
MACS通过整合序列标签位置信息和方向信息提高结合位点的空间分辨率。MACS的工作流如下所示:
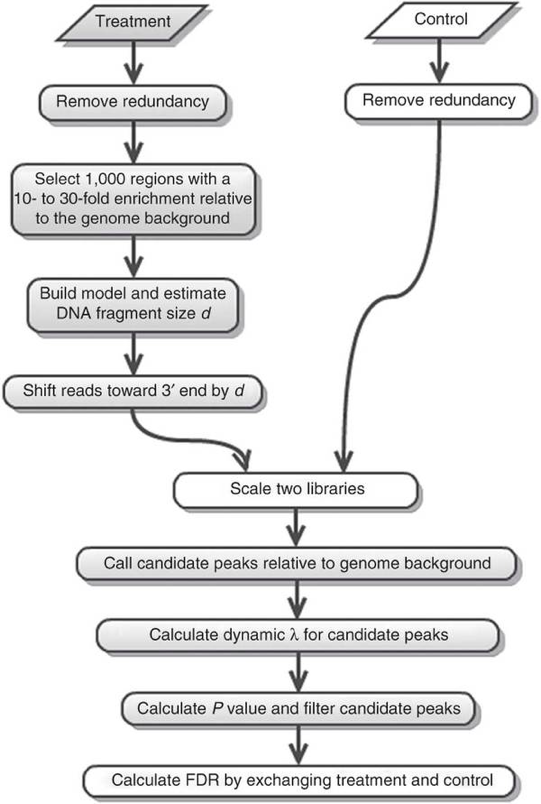
MACS2的使用方法
了解相关参数:
输入文件参数:
-t:实验组,IP的数据文件c: 对照组f:指定输入文件的格式,默认是自动检测输入数据是什么格式,支持bam,sam,bed等g:有效基因组大小,由于基因组序列的重复性,基因组实际可以mapping的大小小于原始的基因组。这个参数要根据实际物种计算基因组的有效大小。软件里也给出了几个默认的-g 值:hs – 2.7e9表示人类的基因组有效大小(UCSC human hg18 assembly).- hs: 2.7e9
- mm: 1.87e9
- ce: 9e7
- dm: 1.2e8
输出文件参数:
--outdir:输出结果的存储路径-n:输出文件名的前缀-B/--bdg:输出bedgraph格式的文件,输出文件以NAME ’_treat_pileup.bdg’ for treatment data, NAME ’_control_lambda.bdg’ for local lambda values from control显示。
peak calling 参数
-q/--qvalue和-p/--pvalue
q value默认值是0.05,与pvalue不能同时使用。--broad
peak有narrow peak和broad peak, 设置时可以call broad peak 的结果文件。--broad-cutoff
和pvalue、以及qvalue相似--nolambda: 不要考虑在峰值候选区域的局部偏差/λq值与峰宽有一定的联系。理想情况下,如果放宽阈值,您将简单地获得更多的peaks,但是使用MACS2放松阈值也会导致更宽的peaks。
Shift 模型参数:
--nomodel
这个参数和extsize、shift是配套使用的,有这个参数才可以设置extsize和shift。--extsize
当设置了nomodel时,MACS会用--extsize这个参数从5’->3’方向扩展reads修复fragments。比如说你的转录因子结合范围200bp,就设置这个参数是200。--shift
当设置了–nomodel,MACS用这个参数从5’ 端移动剪切,然后用–extsize延伸,如果–shift是负值表示从3’端方向移动。建议ChIP-seq数据集这个值保持默认值为0,对于检测富集剪切位点如DNAsel数据集设置为EXTSIZE的一半。
示例:
- 想找富集剪切位点,如DNAse-seq,所有5’端的序列reads应该从两个方向延伸,如果想设置移动的窗口是200bp,参数设置如下:
--nomodel --shift -100 --extsize 200 - 对nucleosome-seq数据,用核小体大小的一半进行extsize,所以参数设置如下:
--nomodel --shift 37 --extsize 73
--call-summits
MACS利用此参数重新分析信号谱,解析每个peak中包含的subpeak。对相似的结合图谱,推荐使用此参数,当使用此参数时,输出的subpeak会有相同的peak边界,不同的绩点和peak summit poisitions.
ATAC-Seq call peaks示例
ATAC-seq关心的是在哪切断,断点才是peak的中心,所以使用shift模型,–shift -75或-100
对人细胞系ATAC-seq 数据call peak的参数设置如下:
- macs2 callpeak -t H1hesc.final.bam -n sample --shift -100 --extsize 200 --nomodel -B --SPMR -g hs --outdir Macs2_out 2> sample.macs2.log
MACS2输出文件解读
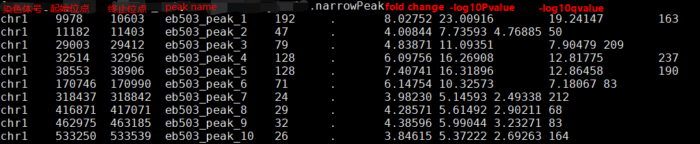
- NAME_peaks.xls
包含peak信息的tab分割的文件,前几行会显示callpeak时的命令。输出信息包含:- 染色体号
- peak起始位点
- peak结束位点
- peak区域长度
- peak的峰值位点(summit position)
- peak 峰值的高度(pileup height at peak summit, -log10(pvalue) for the peak summit)
- peak的富集倍数(相对于random Poisson distribution with local lambda)
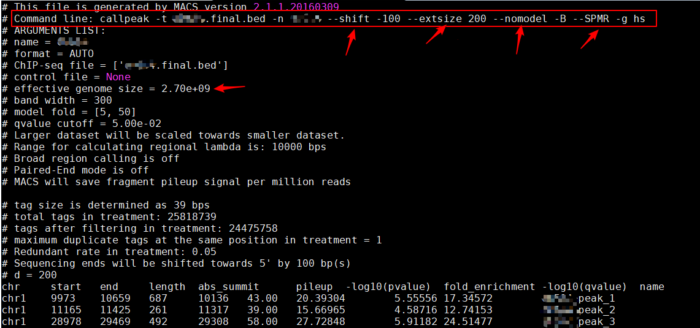
Coordinates in XLS is 1-based which is different with BED format
XLS里的坐标和bed格式的坐标还不一样,起始坐标需要减1才与narrowPeak的起始坐标一样。
- NAME_peaks.narrowPeak
*narrowPeak文件是BED6 4格式,可以上传到UCSC浏览。输出文件每列信息分别包含:- 1;染色体号
- 2:peak起始位点
- 3:结束位点
- 4:peak name
- 5:int(-10*log10qvalue)
- 6 :正负链
- 7:fold change
- 8:-log10pvalue
- 9:-log10qvalue
- 10:relative summit position to peak start(?)
- NAME_summits.bed
BED格式的文件,包含peak的summits位置,第5列是-log10pvalue。如果想找motif,推荐使用此文件。Remove the beginning track line if you want to analyze it by other tools.???
- .bdg
bedGraph格式,可以导入UCSC或者转换为bigwig格式。两种bfg文件:treat_pileup, and control_lambda.
- .bdg
- NAME_peaks.broadPeak
BED6 3格式与narrowPeak类似,只是没有第10列。
summits.bed,narrowPeak,bdg, xls四种类型输出文件的比较:
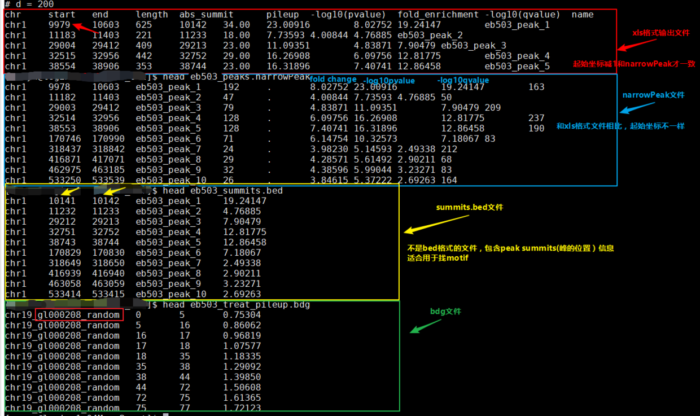
- xls文件
文件包含信息还是比较多的,和narrowPeak唯一不同的是peak的起始位置需要减1才是bed格式的文件,另外还包含fold_enrichment 和narrowPeak的fold change 对应,-log10pvalue,-log10qvalue,peak长度,peak 峰值位置等。 - narrowPeak文件
和xls文件信息类似 - summits.bed文件
包含峰的位置信息和-log10pvalue - bdg文件
bdg文件适合导入UCSC或IGV进行谱图可视化,或者转换为bigwig格式再进行可视化。