

ATAC-Seq分析教程系列
ATAC-Seq分析教程:ATAC-seq的背景介绍以及与ChIP-Seq的异同
ATAC-Seq分析教程:用MACS2软件call peaks
ATAC-Seq分析教程:对ATAC-Seq/ChIP-seq的质量评估(一)phantompeakqualtools
ATAC-Seq分析教程:对ATAC-Seq/ChIP-seq的质量评估(二)ChIPQC
ATAC-Seq分析教程:用ChIPseeker对peaks进行注释和可视化
ATAC-Seq分析教程:用网页版工具做功能分析和motif分析
1. 学习目标
- 讨论ChIP-seq数据质量评估的其他方法
- 用ChIPQC产生质量统计报告
- 鉴定低质量数据的来源
- 概览图
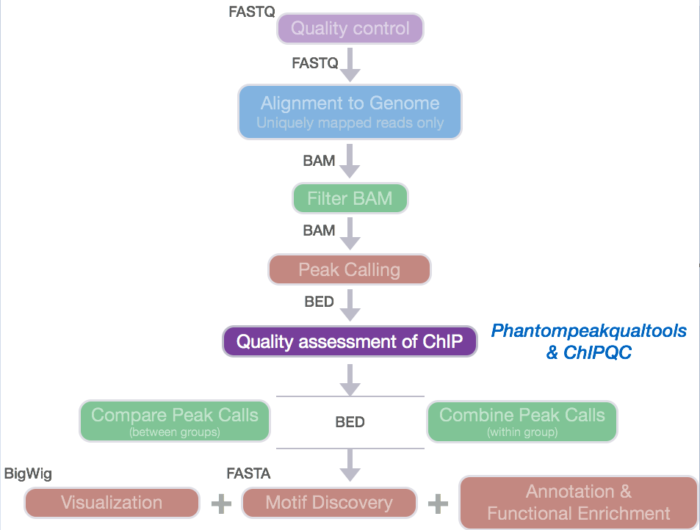
ENCODE评估数据质量采用多种指标,如前面已经讨论过的链相关的指标NSC和RSC。这一节将会讨论评估信号分布的其他指标。
NOTE:这里给出的评估指标只是反映数据质量的好坏,符合阈值的并不意味着实验是成功的,不符合阈值的也不一定意味着失败。
2.常见质量评估指标的介绍
SSD
SSD值是对富集效果的评估。SSD值依赖于全基因组的pile-up信号强度,对真实的ChIP富集和干扰的强信号区域都很敏感。SSD值越大表明富集越好。
“It provides a measure of pileup across the genome and is computed by looking at the standard deviation of signal pile-up along the genome normalised to the total number of reads. ”
FRiP:Fraction of reads in peaks
FRiP表示的是peaks中的reads与总reads的比例。它是另一个反映样本富集效果或IP好坏的评价指标。可以理解为是“信噪比”即文库中结合位点片段占背景reads的比例。一个典型质量好的TF富集FRiP值约5%或者更高,polII的FRiP值约为30%或者更高,也有一些质量好的数据FRiP值<1%(如RNAPIII)
Relative Enrichment of Genomic Intervals (REGI)
REGI是对peaks在不同基因组特征位点分布的统计。
RiBL: Reads overlapping in Blacklisted Regions
过滤人工造成的高信号区域非常重要,如ENCIDE和modENCODE提供的DAC Blacklisted Regions track。这些区域经常在特定的重复序列处出现,如着丝粒、端粒、卫星重复序列等,通过简单的比对过滤是不能去除的。来自blacklisted regions的信号会造成call peak 和片段长度评估的混淆。
RiBL值可以表示背景信号或input的信号水平,与input sample的SSD值以及input和ChIP sample的读长覆盖值相关。这些区域通常是基因组的0.5%,或者更高的比例(10%)。
3. ChIPQC: quality metrics report
ChIPQC是一个Bioconductor包,输入文件包括BAM和peak文件,可以自动计算一些质量评估值,并产生质量报告。
准备数据
- BAM files
首先对比对过滤后的bam数据(chr12_aln.bam)建索引,然后将bam和index文件从~/ngs_course/chipseq/results/bowtie2移动到自己的目录文件夹data/bams - peak files
将narrowPeak 文件从macs2目录下~/ngs_course/chipseq/results/macs2移动到自己目录下data/peakcalls - sampleSheet file
sampleSheet file是唯一需要自己根据实验设计和数据存储地址等信息创建的一个csv格式文件(bam,peak文件分别在比对和call peak的步骤产生)。sampleSheet具体需要包含的信息如下:

- SampleID: 样本ID
- Tissue, Factor, Condition: 不同的实验设计对照信息,三列信息必须包含在sampleSheet里,如果没有某一列的信息设为NA。
- Replicate : 重复样本的编号
- bamReads : 实验组BAM 文件的路径(data/bams)
- ControlID : 对照组样本ID
- bamControl :对照组样本的bam文件路径
- Peaks :样本peaks文件的路径
- PeakCaller :peak类型的字符串,可以是raw,bed,narrow,macs等。
下载安装ChIPQC
- source("http://bioconductor.org/biocLite.R")
- biocLite("ChIPQC")
Running ChIPQC
ChIPQC只需要三步就可以完成质量评估和报告生成。
- 首先载入包和sampleSheet信息
- ## Load libraries
- library(ChIPQC)
- ## Load sample data
- samples <- read.csv('meta/samplesheet_chr12.csv')
- View(samples)
- 创建ChIPQC对象
利用sampleSheet的信息读取每个样本的bam和narrowpeak文件,并计算质量评估值,结果存在一个对象里。- ## Create ChIPQC object
- chipObj <- ChIPQC(samples, annotation="hg19")
- 生成ChIPQC报告
- ## Create ChIPQC report
- ChIPQCreport(chipObj, reportName="ChIP QC report: Nanog and Pou5f1", reportFolder="ChIPQCreport")
ChIPQC报告解读
ChIPQC生成的结果包含一个网页报告和报告中含有的所有图片。
网页报告有三部分:QC Summary ;QC Results;QC files and versions

(1)QC Summary - Overview of results
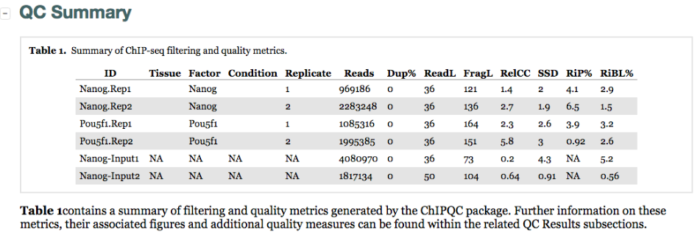
QC summary包含sampleSheet里填写的样本的基本信息Tissue,Factor,Condition,Replicate。另外还有上面提到的质量评估的常用指标SSD、RiP%和RiBL值。越高的SSD值表明富集效果越好,Pou5f1样本(2.6,3)有较高的SSD值,RiBL值不是很高,FRiP的比例在5%附近或者更高,除了Pou5f1-rep2。
- SSD - SSD score (htSeqTools)
- RIP% - Percentage of reads wthin peaks
- RIBL% - Percentage of reads wthin Blacklist regions
同时表格中还给出了其他统计信息: - Reads - Number of sample reads within analysed chromosomes.
- Dup% - Percentage of MapQ filter passing reads marked as duplicates
- FragLen - Estimated fragment length by cross-coverage method
- FragLenCC - Cross-Coverage score at the fragment length
- RelativeCC - Cross-coverage score at the fragment length over Cross-coverage at the read length
(2)QC Results - Full QC results and figures
- Mapping, Filtering and Duplication rate
第一部分是比对、过滤和重复率质检结果,包括Table2 、Figure1和Figure2。
Table 2主要给出了比对质量和重复率,因为BAM文件是过滤后的,所以这里Dup%都是0.
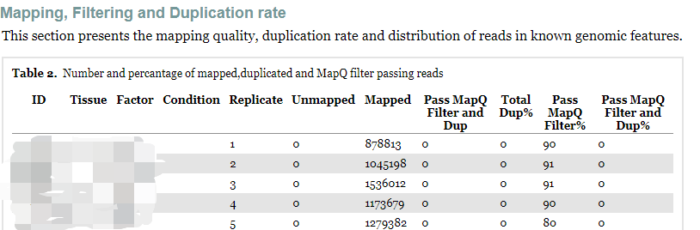
- Total Dup%-Percentage of all mapped reads which are marked as duplicates.
- Pass MapQ Filter%-Percentage of all mapped reads whichpass MapQ quality filter
- Pass MapQ Filter and Dup%-Percentage of all reads which pass MapQ filter and are marked asduplicates.
Figure 1展现了reads在blacklists中的比例,
Figure 2是用基因组注释呈现了reads在基因组特征位置如启动子的分布。这幅图里显示在启动子区域富集最明显。
- ChIP signal Distribution and Structure
第二部分是ChIP信号分布和结构组成,包括Figure3和4。
Figure 3是一个coverage plot, x轴代表在某bp位置read pileup的高度,y轴代表有多少位置有相同的pileup 高度(取log)。**有好的富集的ChIP样本会有一个tail,即更多的位置(y值大)有较高的测序深度。在我们的数据集中Nanog样本与Pou5f1 相比有较高的tails,尤其是重复样本2。但是Pou5f1有较高的SSD值。当SSD高但是coverage看起来低时,可能是存在大片段深度高的区域出现在blacklist 基因组区域。 - Peak Profile and ChIP Enrichment
第3部分是peak的谱图和ChIP的富集,每个peak都集中在summit位置(summit 理解为peak的最高峰值点处)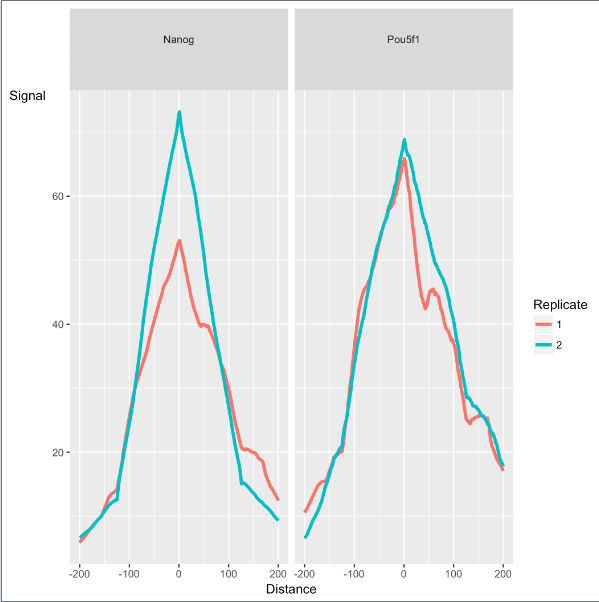
- peak的性状取决于研究对象的类型,如转录因子、组蛋白标记、或其他DNA结合蛋白如聚合酶等,相同类型的对象通常有独特特征的谱图。
Figure6和7都是对比对到peak中的reads统计。富集效果好的ChIP样本的reads与peaks会有高比例的重合。尽管Nanog有较高的RiP,但是两个重复样本间的差异大于Pou5f1。 - Figure8和9**表示样本的聚类效果,分别是相关性聚类热图和PCA。
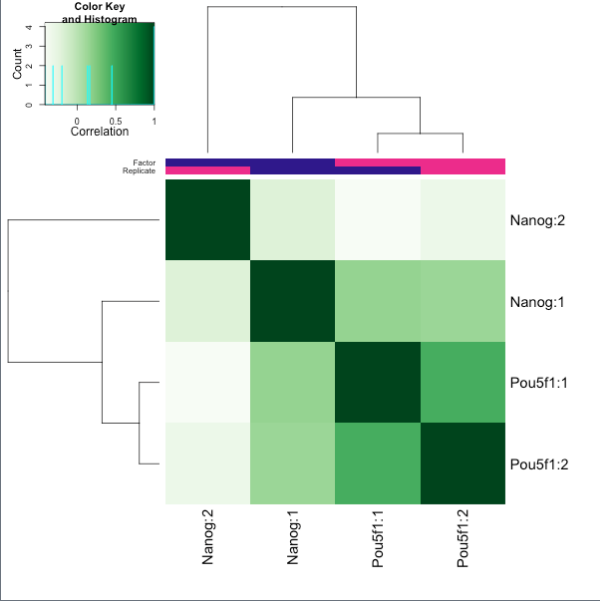
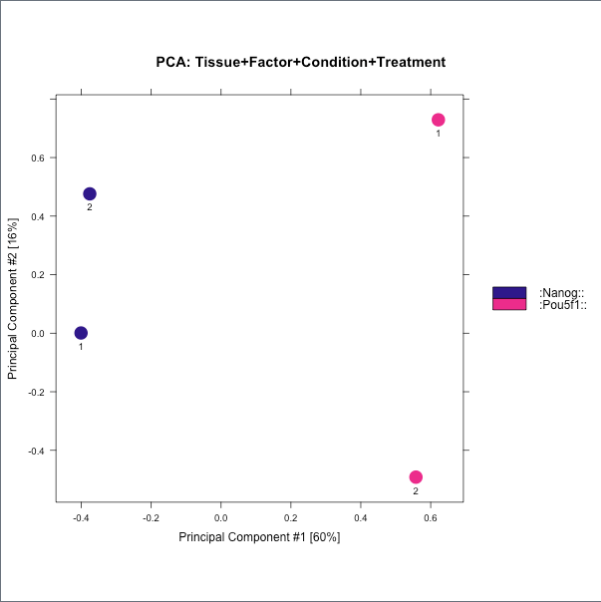
4. 实验偏差:ChIP-seq数据质量低的来源
- 免疫沉淀的特异性和有效性
影响因素如抗体的特异性,结和沉淀的强度 - 片段化
超声裂解产生不同大小的片段可能引入偏差 - 文库构建时的偏差
如PCR扩增