

1.Hi-C原理简介
- 1.1 Hi-C技术高通量染色体构象捕获技术(
High-throughput chromosome conformation capture)研究全基因组三维构象及分析染色质片段相互作用的实验技术 - 1.2 Hi-C目的了解核内染色质的三维构象、获得细胞核内空间位置非常接近或存在相互作用的染色质测序片段更好地研究染色质内或染色质间的互作、基因调控元件在全基因组范围内调控的情况
- 1.3 Hi-C应用方向辅助基因组组装、揭示空间调控、揭示物种进化、疾病研究、三维结构差异分析、还原染色体三维结构、构建染色体跨度单体型
- 1.4 互作本质统计学上基因组两点之间发生空间接触的概率
- 1.5 Hi-C实验原理
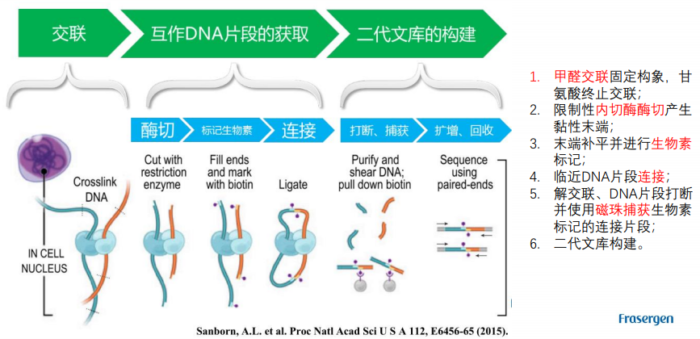 Hi-C实验原理
Hi-C实验原理 - 1.6 二代文库构建及测序二代文库进行片段筛选400-600bp的片段,实际插入片段长度为300-500bp一般测序读长:PE150
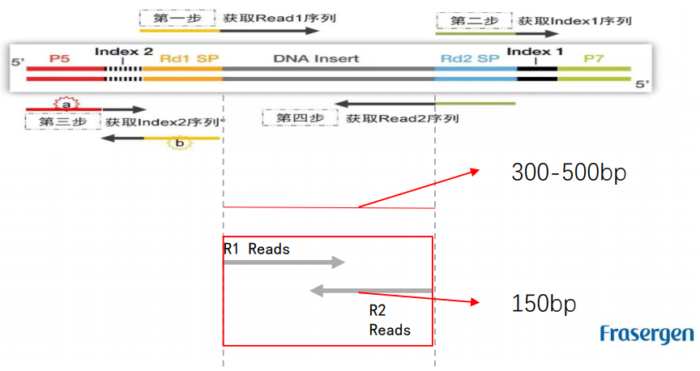 二代测序
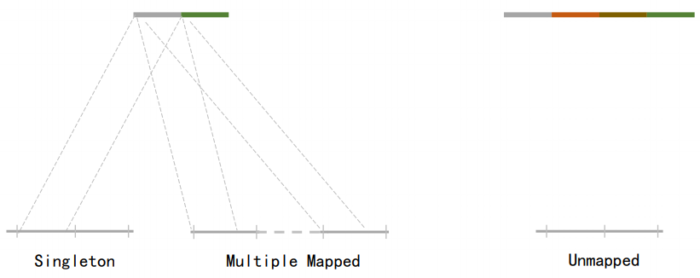
二代测序 - 1.7 Hi-C实际文库类型将HIC数据进行比对是会出现不同的比对情况,我们需要的是双端唯一匹配。对单端匹配、多处比对、未比对的reads进行过滤。
对Hi-C文库构建中产生的多种分子类型,包括 re-ligation、Dangling ends、self circle 、dump reads 及valid pairs reads等类型。 在 Hi-C 分析中,仅valid pair可以反映基因组上位点与位点间的互作信息。因此,
非重复的valid pair所占的比例是评估Hi-C文库质量的 重要指标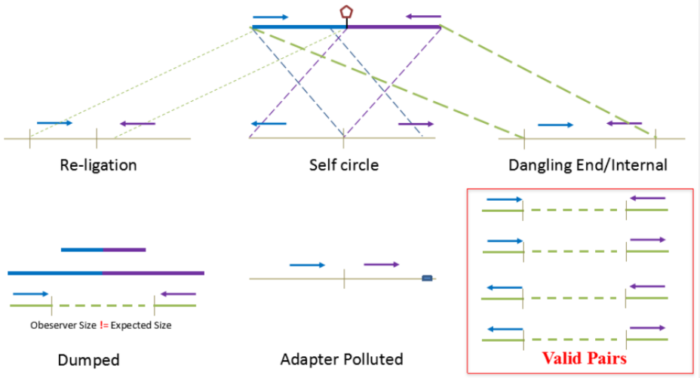
- 互作矩阵的生成由于计算资源,数据量等因素,我们往往认为确定一个互作单位:
bin。将基因组按照一定大小分成bin。将过滤后的有效序列分配到这些bin中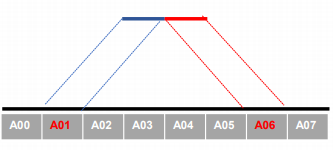
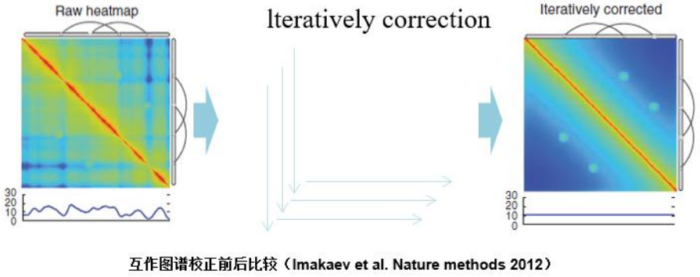
- 互作矩阵的矫正Hi-C数据中由于内切酶的偏好性、基因组本身质量、基因组序列特异性会导致其在基因组不同位置间存在偏差。因此,我们会对互作矩阵进行校正,使其数据在基因组上每个位点的覆盖度一致。常用的矫正方式有迭代矫正、归一化等
2.比对软件介绍
- 常用短序列比对软件
| Bowtie2 | BWA | |
|---|---|---|
| 算法原理 | FM-Index(基于BWT) | BWT construction algorithm |
| 常用比对模式 | End-to-End | Mem(pair-end) |
| 输出 | SAM、TSV | SAM |
| 特点 | 支持单端、双端reads比对;支持插入、缺失错误比对 | 支持单端、双端reads比对;支持插入、缺失、嵌合reads比对 |
| 区别 | MAPQ值打分算法不同于BWA | 处理嵌合reads时会分段输出比对结果;基因组mapping率略高于Bowtie2 |
- SAM格式详解SAM分为两部分,注释信息(header section)和比对结果部分(alignment section)注释信息:可有可无,以
@开头,用不同的tag代表不同的信息比对结果:
| 列 | 字段名 | 中文解释 | 举例 |
|---|---|---|---|
| 1 | QNAME | 比对片段的编号,read name | V300059328L4C001R0010000044 |
| 2 | FLAG | 位标符,reads mapping情况的数字表示 | 16 |
| 3 | RNAME | 比对上参考序列的编号 | chr10 |
| 4 | POS | 比对上参考序列的位置,1-based | 321541 |
| 5 | MAPQ | 比对的质量分数MAPQ=-10 * log10(mapping出错的概率) | 60 |
| 6 | CIGAR | 简要比对表达式 | 150M |
| 7 | MRNM | mate比对上的参考序列 | chr10 |
| 8 | MPOS | mate比对参考序列的位置 | 322000 |
| 9 | ISIZE | reads比对长度 | 470 |
| 10 | SEQ | reads的序列 | |
| 11 | QUAL | ASCII 码格式的序列质量 | |
| 12 | 可选区域 | AS:i 匹配的得分;XS:i 第二好的匹配的得分;YS:i mate 序列匹配的得分 |
3.HiC常规软件比较
| 软件名 | hiclib | HiC-Pro | HICUP | Juice |
|---|---|---|---|---|
| 比对软件 | Bowtie2 | Bowtie2 | Bowtie2 | BWA-mem |
| 比对策略 | 迭代比对 | 全局、局部比对 | 先截短后比对 | Pair-end,嵌合reads过滤 |
| 嵌合reads处理 | √ | √ | √ | √ |
| 构建矩阵 | √ | √ | × | √ |
| 标准化 | ICE | ICE | × | KR |
| 结果文件 | hdf5、hm、bychr(HDF5) | SAM、validpair | SAM | SAM、MND、.hic |
| 特点 | 比对结果可靠,存储消耗小 | 简单易用,输出结果可读 | 过滤非常严格 | 后续分析接口多,juicebox可视化 |
4.HiC-Pro代码实操
4.1 软件安装
- HiC-Pro软件安装(需要的包有点多,些许繁琐)
- git clone https://github.com/nservant/HiC-Pro.git
- cd ./HiC-Pro
- vi config-install.txt
- 修改HiC-Pro目录下的config-install.txt
- #########################################################################
- ## Paths and Settings - Start editing here !
- #########################################################################
- PREFIX = 文件安装位置
- BOWTIE2_PATH = bowtie2安装目录
- SAMTOOLS_PATH = samtools安装目录
- R_PATH = R的安装目录
- PYTHON_PATH = python安装目录
- CLUSTER_SYS = 用于集群提交的调度器,必须为TORQUE,SGE,SLURM,LSF四个中的一种
- 修改保存后
- make CONFIG_SYS=config-install.txt install
4.2 bowtie2索引构建
- bowtie2-build [options] <reference> <bt2_index_base>
reference : 下载的参考基因组,genome.fa
bt2_index_base: 构建索引前缀
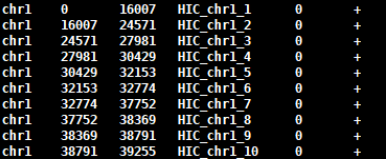
4.3 使用digest_genome.py生成酶切片段文件
python HiC-Pro/bin/utils/digest_genome.py -r [常用限制性内切酶序列] [-o OUT] fastafile
-r:常用限制性内切酶:
| 限制性内切酶 | 酶切位点,^为切割位点 |
|---|---|
| MboI | ^GATC |
| DpnII | ^GATC |
| BglII | A^GATCT |
| HindIII | A^AGCTT |
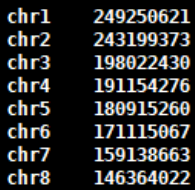
4.3 生成基因组sizes文件,获得基因组每条染色体bases数bed文件
- samtools faidx genome.fa
- awk ‘{print $1 "t" $2}‘ genome.fa.fai > genome_sizes.bed
4.4 Hi-C数据准备
- 创建sample文件夹,一个文件夹放入一个样品的fastq文件(生物学重复可以放入)

4.5 配置Config文件
vi ./config-install.txt
- 需要修改的参数有:
N_CPU:给定的CPU内存数,给的越多,运行的越快(根据服务器配置);LOGFILE:日志文件的名称;JOB_MEM:内存的大小PAIR1_EXT= _R1:R1测序数据名称中有_R1PAIR2_EXT = _R2:R2测序数据名称中有_R2MIN_MAPQ: 最低的质量分数,用于筛选,表示低于该MAPQ值会被过滤BOWTIE2_IDX_PATH: 基因组bowtie2索引路径,eg:/path/hg19BOWTIE2_GLOBAL_OPTIONS: 默认GLOBAL比对设置BOWTIE2_LOCAL_OPTIONS: 默认LOCAL比对设置REFERENCE_GENOME: Bowtie2索引前缀GENOME_SIZE: 基因组sizes bed文件GENOME_FRAGMENT: 基因组酶切文件,eg. /path/hg19_HindIII.bedLIGATION_SITE: 酶切位点末端补平再次连接后形成的嵌合序列,eg. AAGCTAGCTTMIN_FRAG_SIZE: 最小的理论酶切片段大小,eg. 100MAX_FRAG_SIZE: 最大的理论酶切片段大小,eg. 100000MIN_INSERT_SIZE: 最小的文库片段大小,eg.100MAX_INSERT_SIZE: 最大的文库片段大小,eg.1000BIN_SIZE:需要生成的矩阵分辨率(bp)MATRIX_FORMAT:矩阵的形式,upper表示保留上半部分
4.6 HiC-Pro运行
HiC-Pro -i INPUT -o OUTPUT -c CONFIG [-s ANALYSIS_STEP] [options]
-c: config文件路径
-o: 结果生成路径
-i: 原始数据路径
-p: 集群运行
5.结果解读
- 总目录
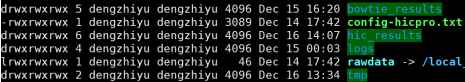
bowtie_results:比对结果目录
hic_results:hic矩阵及分析结果目录
logs:存放分析日志
rawdata:链接了原始数据
tmp:存放中间文件
- Bowtie_result目录
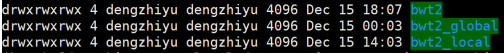
bwt2:存放合并后的bam文件和统计结果
bwt2_global:存放全局比对结果
bwt2_local:存放局部比对结果
- hic_result目录
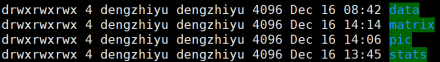
data:存放validpair及其他无效数据文件
matrix:存放不同分辨率矩阵文件
pic:存放统计分析图片
stats:存放统计表
- Data文件
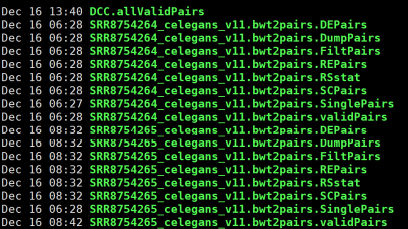
allVaildPairs:合并后的pairs数据
DEPairs:Dangling end pairs数据
DumpPairs:实际片段长度和理论片段长度
不同的数据
REPairs:酶切片段重新连接的pairs
FiltePairs:MAPQ过低的pairs
SCPairs:片段自连的pairs
- Matrix文件
raw:原始矩阵iced:ice标准化后的矩阵 - Pic文件,出图
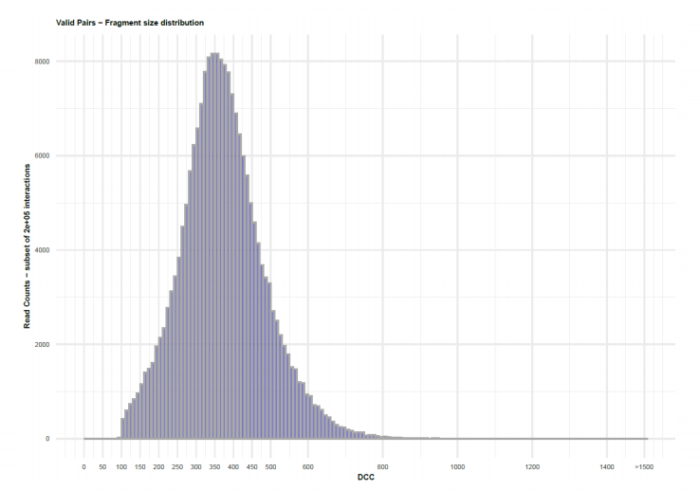 HiC文库片段分布文件
HiC文库片段分布文件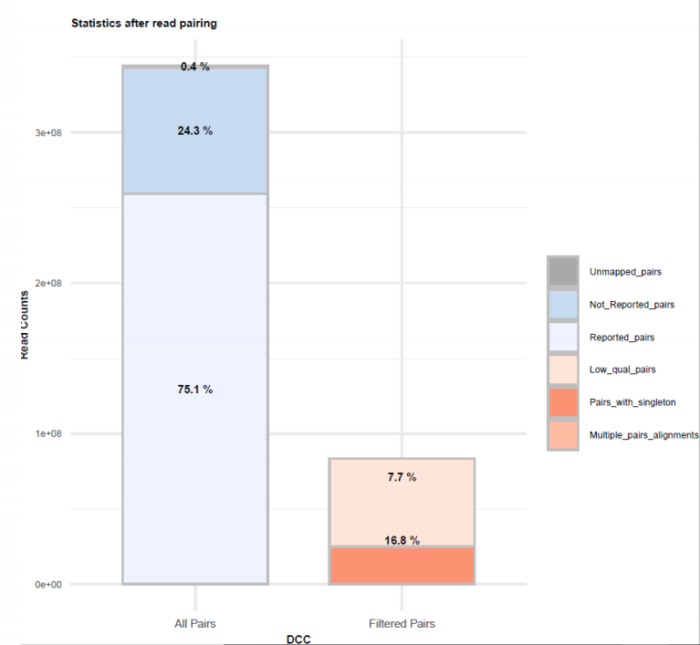 双端比对过滤质控图
双端比对过滤质控图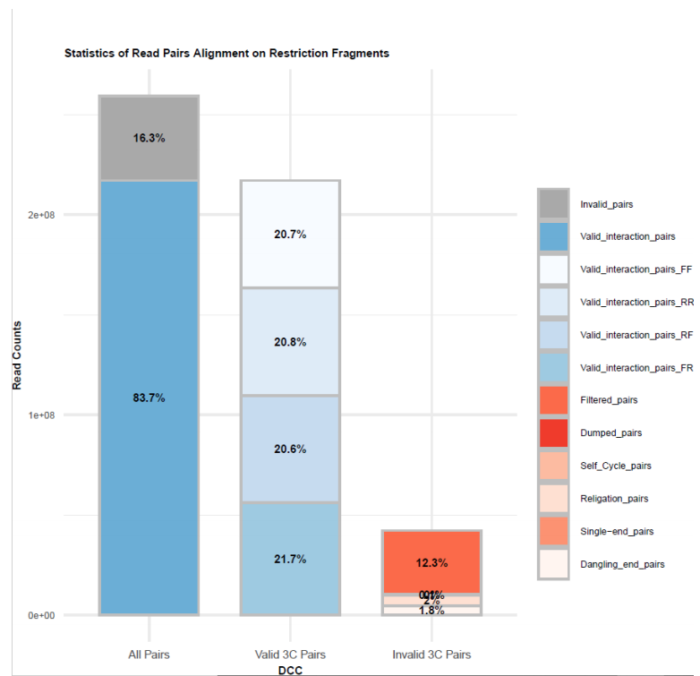 有效数据过滤质控图
有效数据过滤质控图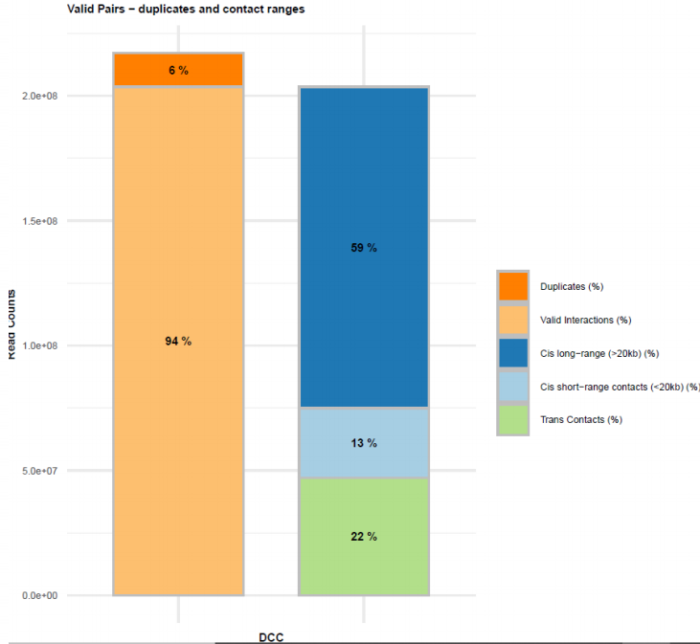 配对数据不同类型数据比例展示图
配对数据不同类型数据比例展示图