

在科学研究中,设立对照是一项基本原则,如病例对照研究的病例组和对照组、队列研究中的暴露组和非暴露组,临床随机对照试验的试验组和对照组。
对这些研究进行Meta分析时合并的指标是两组的相对效应如OR值、RR值或是绝对效应如危险度差值(risk difference,RD),然而在并未设立对照组如流行病学中的现况研究.如要了解某种病毒在全国的一个总体感染率而又没有足够的时间或经费做全国性的调查时.我们可以通过对现有文献报道的感染率进行Meta分析了解该病毒在全国的感染情况。
单个率可包括流行病学现况研究中的患病率、感染率,临床试验的有效率,药物试验中的不良反应发生率等,只要收集到各个原始研究的样本量和事件发生数,在R软件就可以用metaprop 函数对单个率进行定量的Meta分析.来计算合并的率及95%可信区间。通过观察图形是否对称初步判断有无发表偏倚,结合metabias可以绘制出Begg和Egger图, 对发表偏倚进行统计学检验。考虑到不同类型的单个率的资料的分布可能会有不同的情况,R软件给出了五种估计率的方法。如原始率不服从正态分布,可经过转换使其服从或接近正态分布,从而提高合并结果的可靠性。
单组率的meta分析
定义:只提供一组人群的总人数和时间发生人数,多为患病率,检出率,知晓率,病死率,感染率。一般基于的研究为横断面研究。
缺点:异质性很难控制
方法:①加权计算:即根据每个独立研究的样本量大小,给予不同的权重,对各独立样本的效应量率进行合并; ②直接等权相加: 即把各独立的结果事件直接等权相加,然后直接计算合并率,再用近似正态法计算其可信区间 ; ③调整后再等权相加: 即对各个独立研究资料的率进行调整后再行等权相加,计算出合并率的大小。
Meta analysis软件是一款免费的软件且可以进行单个率的Meta分析.但是其为菜单操作,无法实现对原始率的转换。R软件是一种共享的免费统计软件,有专门的Meta分析程序包,可以进行单个率的Meta分析,而且提供了五种方法估算率,研究者可以根据原始率的分布选择合适的方法。本文结合编程和Meta分析程序包,以实例说明R软件在单个率Meta分析中的应用,以期为今后的Meta分析提供方法学指导。
具体操作
1.加载meta数据包
library(meta)
2.具体分析操作
数据的读取
> library(meta)
> setwd('/home/ub/Rwork')
> meta_data <- read.csv('metadata.csv',header = T) > head(meta_data)
Author Year Case Number
1 Chen_et 2005 8 40
2 Beyer_et 2001 12 32
3 Ber_et 2003 10 50
4 Loe_et 2010 103 374
5 Alis_et 2011 20 69
6 Choi_et 2015 29 104
这是生成的一份记录疾病患病率的Meta分析。其中变量 Author Year Case Number分别表示文献的第一作者,发表年份,患者和调查的总人数和。
样本率的估计方法的选择
单个率资料的Meta分析要求率的分布应该尽量的服从正态分布。如原始率不服从正态分布,可经过转换使其服从或接近正态分布,从而提高合并结果的可靠性。命令metaprop()进提供了5种样本率的估计方法,根据样本率的分布决定使用哪种合并方法,五种估计方法如下:“PRAW”(没有转换的原始率), ”PLN”(对数转换), ”PLOGIT“(logit转换), “PAS”(反正弦转换),“PFT“(Freeman-Tukey双重反正弦转换), 在进行Meta分析之前,对原始率及按四种估计方法进行转换后的率进行正态性检验,根据检验结果选择最接近正态分布的方法。具体的命令:
rate <- transform(meta, p = Case/Number, log=log(Case/Number), logit=log((Case/Number)/(1-Case/Number)), arcsin.size=asin(sqrt(Case/(Number 1))), darcsin=0.5*(asin(sqrt(Case/(Number 1))) asin((sqrt(Case 1)/(Number 1))))) shapiro.test(rate$p) shapiro.test(rate$log) shapiro.test(rate$logit) shapiro.test(rate$arcsin) shapiro.test(rate$ darcsin) shapiro.test(rate$p) Shapiro-Wilk normality test data: rate$p W = 0.89359, p-value = 0.3374 > shapiro.test(rate$log)
Shapiro-Wilk normality test
data: rate$log
W = 0.89239, p-value = 0.3309
> shapiro.test(rate$logit)
Shapiro-Wilk normality test
data: rate$logit
W = 0.89524, p-value = 0.3466
> shapiro.test(rate$arcsin)
Shapiro-Wilk normality test
data: rate$arcsin
W = 0.89211, p-value = 0.3294
> shapiro.test(rate$ darcsin)
Shapiro-Wilk normality test
data: rate$darcsin
W = 0.88822, p-value = 0.309
结果显示原始率,对数转换, logit转换, 反正弦转换, Freeman-Tukey双重反正弦转换的正态性的结果分别是W = 0.89359, p-value = 0.3374;W = 0.89239, p-value = 0.3309 ;W = 0.89524, p-value = 0.3466; W = 0.89211, p-value = 0.3294;W = 0.88822, p-value = 0.309, 综上,我们选择双重反正弦转换,当然也可以试着选择其他的转换类型, 然后通过比较异质性的大小,选择一个合适的类型。
效应量的合并
通过命令metaprop(),进行率的合并。具体的命令:
meta1 <- metaprop (Case ,Number, data=meta, studlab=paste(Author),sm="PFT")
meta1
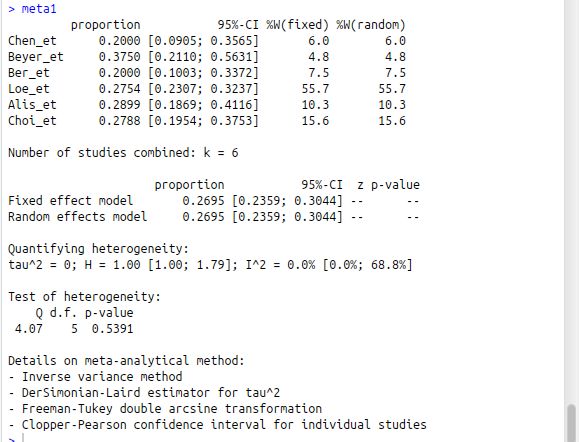
结果显示,异质性检验Q=4.07 P<0.539 I2=0,因此 认为没有统计学意义上的异质性,所以优先选用固定效用模型,如果I2较大,说明6个原始研究间数据存在一定的异致性,则选用随机效应模型。
再看合并的率患病率= 0.1893, 95% CI: 0.1175-0.2736。
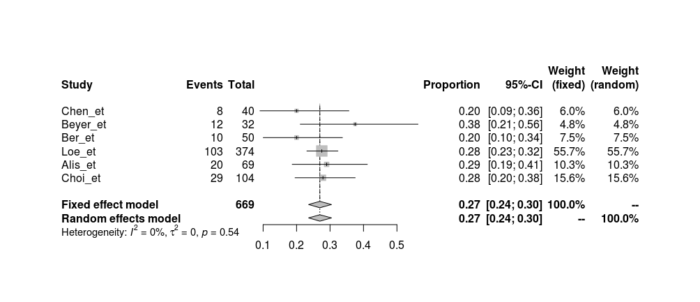
下图所示为Meta分析的森林图。
森林图的绘制
具体的命令如下:forest(meta1)
从森林图中,非常简单和直观地看到Meta分析的统计结果
发表偏倚的检测:运用Egger检验检测发表偏倚,发表偏倚的命令是metabias()。具体的命令:
metabias(meta1,k.min=6)
Linear regression test of funnel plot asymmetry
data: meta1
t = -0.17011, df = 4, p-value = 0.8732
alternative hypothesis: asymmetry in funnel plot
sample estimates:
bias se.bias slope
-0.1593819 0.9369520 0.5569597
从结果中显示,t = -0.17011, p-value = 0.8732,因此认为不存在发表偏倚,
结论
Meta分析的结果显示:该病患者患病率为为0.2695 ,95%可信区间为[0.2359; 0.3044] 。
其他
模型选择的标准
若各原始研究间存在异质性,则使用随机效应模型;否则,则使用固定效应模型。
异质性的判断
Meta分析时,若I2<25%,则认为不存在异致性;若I2介于25%-50%之间,则认为异致性程度较小;若I2的值介于50%-75%之间,则认为存在一定的异致性;若I2>75%,则认为存在较大的异致性。
总结
本文结合实例,介绍了在R软件中如何实现单个率的资料Meta分析,由于单个率的Meta分析各原始文献为单个组的率,稳定性可能不同于具有两个组的研究,因此在合并时统计学异质性可能会比较大,当异质性较大时,首先要从专业性的角度对不同情况下的率进行亚组分析等来确定Meta分析的结果是否可靠。