

关于批量相关性分析,我们发过两个帖子。单基因批量相关性分析的妙用,又是神器!基于单基因批量相关性分析的GSEA。两两分析的肯定也是没有问题:
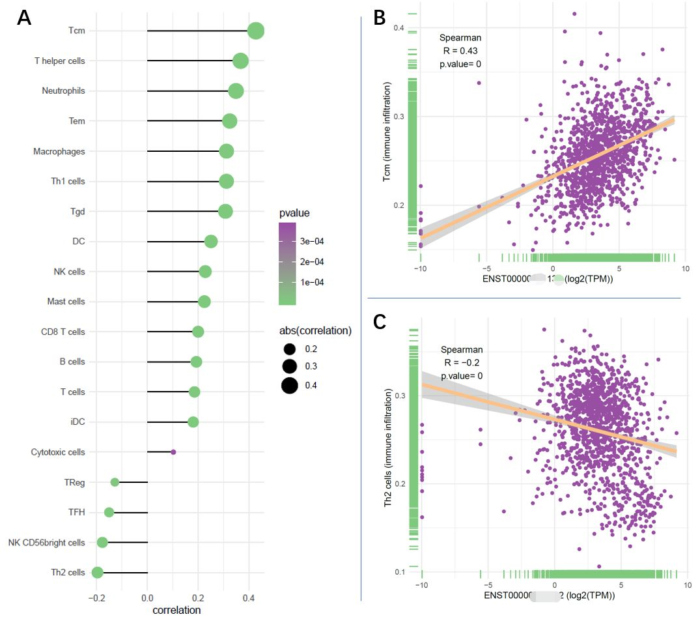
现在的问题是,如果是多个基因分相关性分析,如何快速,方便地分析,然后高效地呈现呢?
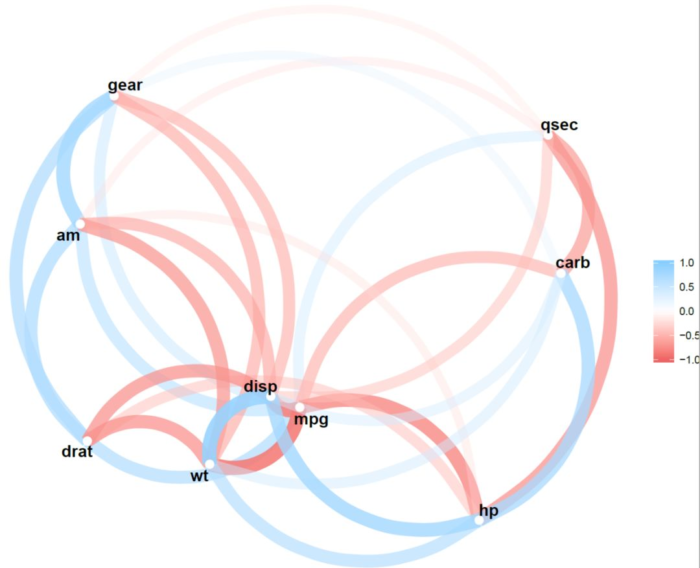
我用圈图实现过这个操作:
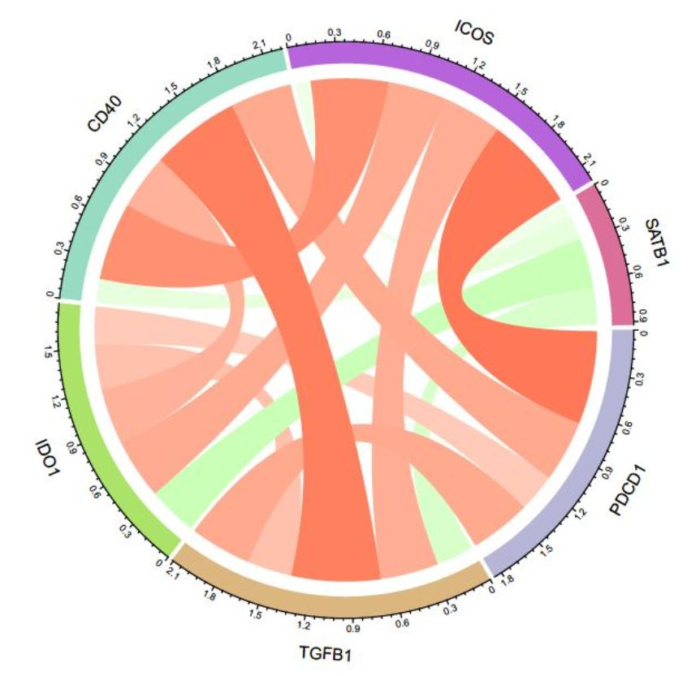
不过比较麻烦,今天介绍一个R包 corrr,可以方便地做这个事情,而且我认为做的更好。
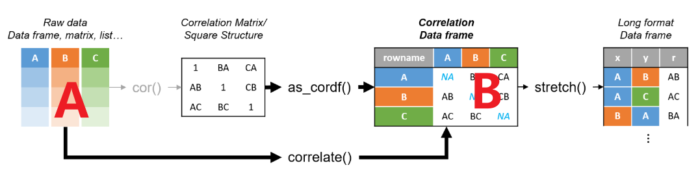
他有一个主函数correlate可以迅速地分析,实现从A到B的转换。
更重要的是他还配了两个可视化的函数,一个是rplot画热图,一个是network_plot画网络图。
出了主函数外,总共有7个函数

最后一个框里有两个函数已经介绍,还有一个fashion可以简洁化展示数据,去掉NA。
第一个框内的shave函数是把剃刀,可以去掉相关性结果上三角或者下三角并设置为NA
rearrange函数可以按照相关性系数聚类排序。
第二个框内的focus函数,类似于select函数,可以用来筛选想要查看的某行某列数据。
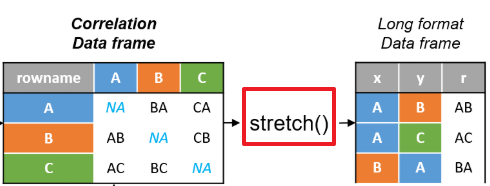
stretch函数可以实现数据从宽边长,如图所示。
下面我们就来实战一下:
首先设置镜像以及按照R包
options("repos"=c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
install.packages("corrr")
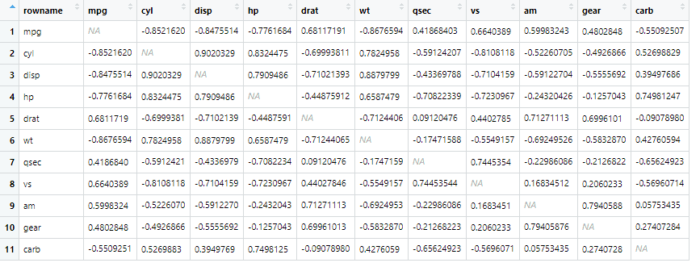
运行主函数看看结果
library(corrr)
library(dplyr)
x <- datasets::mtcars %>%
correlate()
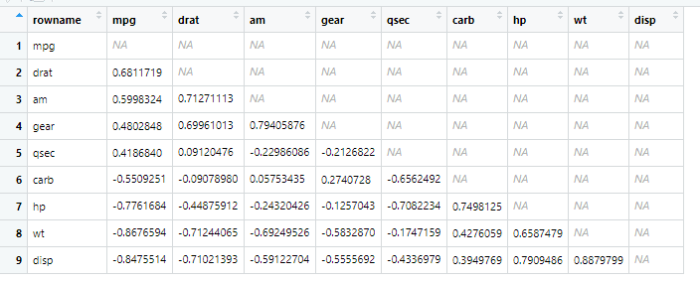
接着往下处理,focus选择要观察的数据,rearrange按照相关性系数排序,shave设置上三角的数据为NA
x <- datasets::mtcars %>%
correlate() %>%
focus(-cyl, -vs, mirror = TRUE) %>%
rearrange() %>%
shave()
可以用fashion简化数据
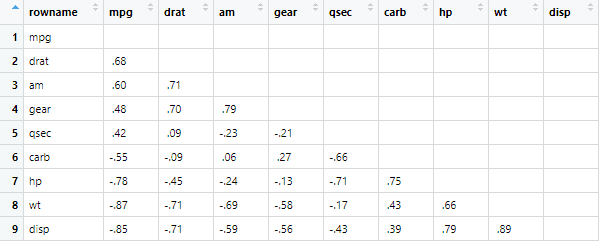
fashion(x)
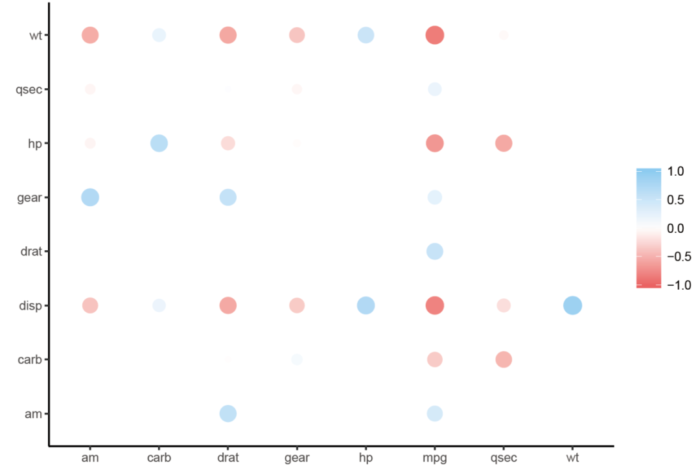
可以用rplot来画图展示数据
也可以用网络图来展示
datasets::mtcars %>%
correlate() %>%
focus(-cyl, -vs, mirror = TRUE) %>%
rearrange() %>%
network_plot(min_cor = .2)
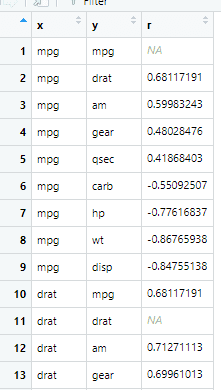
stretch可以实现数据变换
x <- datasets::mtcars %>%
correlate() %>% # Create correlation data frame (cor_df)
focus(-cyl, -vs, mirror = TRUE) %>% # Focus on cor_df without 'cyl' and 'vs'
rearrange() %>%
stretch()
学习一个R包是第一步,第二步就应该想着如何用来展示自己的数据。先来看看我们拥有的数据
load(file = "TCGA_steal_data.Rdata")
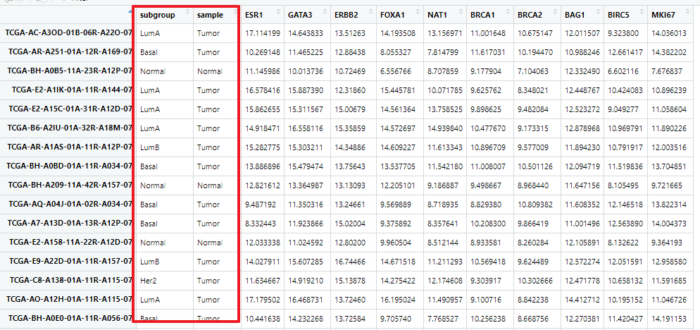
这个数据我们已经很熟悉了,前面两列可以不要
data <- TCGA_steal_data[,-c(1:2)]
这样我们就跟这个R包对接上了。
直接画图试试
data %>%
correlate() %>%
rearrange() %>%
network_plot()
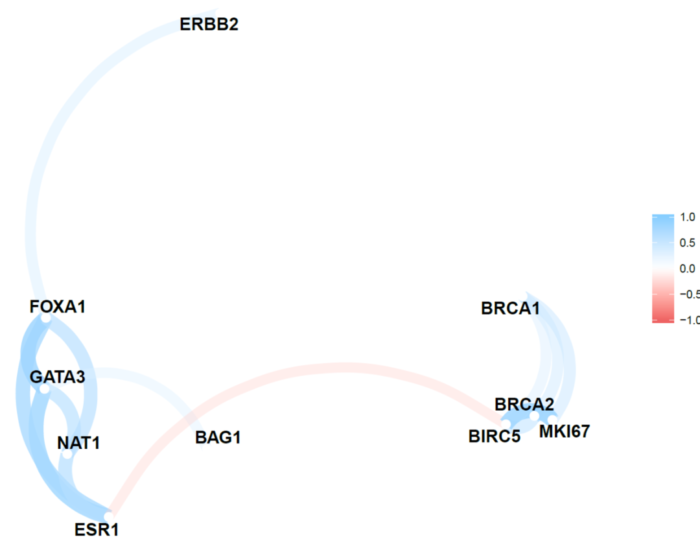
这个图很有意思,我们看到总体分为两群,其中一群是FOXA1,GATA3,ESR1,这是乳腺癌的数据,这三个能聚在一起是符合背景的,因为这三个分子可以决定luminal A型。不过我不知道的是NAT1这个基因跟他们关系这么密切。
另外一群是BRCA1,BRCA2,MKI67,他们之间是正相关,这个可以查一下文献,看看是否是这个样子。
correlate有参数可以限定相关性分析的方法,network_plot也有参数可以设置最终的颜色,以下我给大家展示一下我的洋葱配色
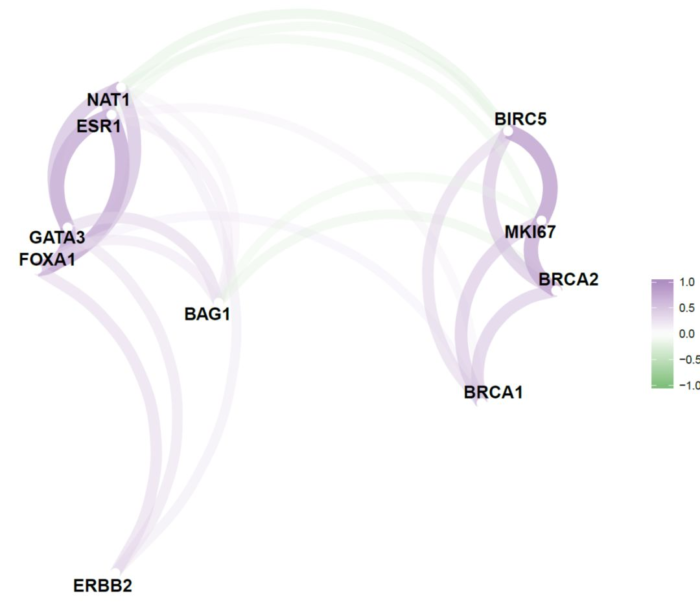
说实话,我挺喜欢这个图的。