

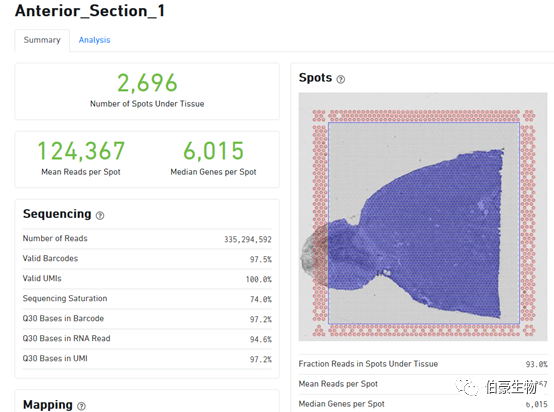
Q30 Bases in UMI:UMI 序列的 Q30 值
Mapping 结果统计区:
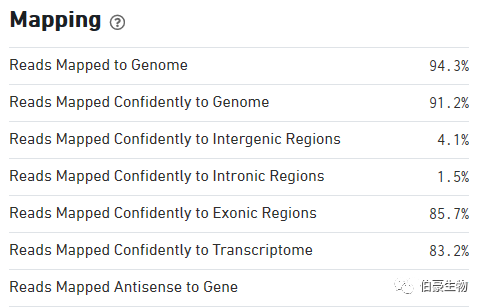
Reads Mapped to Genome:比对到基因组上 reads 的比例
Reads Mapped Confidently to Genome:唯一比对到基因组上 reads 的比例,也就是我们常说的 mapped uniquely reads,不过这里如果某条 reds 唯一比对到一个基因的 exon 区,同时又比对到了一处非 exon 区,还是算唯一比对到 exon 区的 reads。
Reads Mapped Confidently to Intergenic Regions:比对到唯一基因间区的 reads 的比例
Reads Mapped Confidently to Intronic Regions:比对到唯一内含子区的 reads 的比例
Reads Mapped Confidently to Exonic Regions:比对到唯一外显子区的 reads 的比例
Reads Mapped Confidently toTranscriptome:比对到唯一基因转录组上 reads 的比例,这一部分会包括剪切位点的 reads。这一部分的 reads 就是用来对 UMI 进行计数统计的。细心的朋友可能会发现这一部分的 reads 比例比 Reads Mapped Confidently to Exonic Regions 的值要低,这是因为有些基因的 exon 是有 overlap 的,处于 overlap 区域的 reads 最终是不进入 UMI 计数的。
Reads Mapped Antisense to Gene:比对到基因转录组的反义链区域的 reads 比例,这部分 reads 是没有意义的。从这里我们也可以发现 10x 空间转录组建库和比对有方向性的。
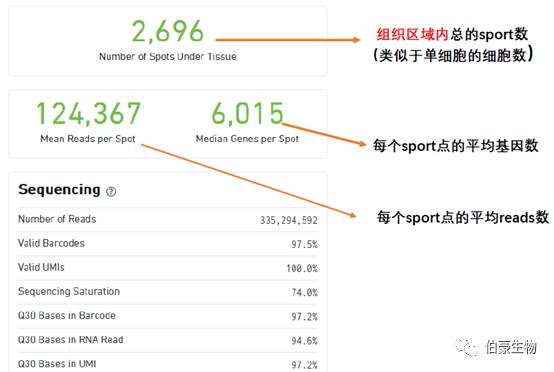
Spot 信息统计区:

Fraction Reads in Spots Under Tissue:比对到唯一基因转录组上 reads(Reads Mapped Confidently to Transcriptome)有多少比例覆盖在组织区域的 spot 上,这里是 93%,那就说明只有 7% 的 reads 分布在组织之外的灰色区域的。10x 软件在这里有一个默认的阈值为 50%,认为这个比例值超过 50% 结果是正常的,低于 50% 则回到网页最上面区域提示报错信息(认为可能是透化不完全导致背景 RNA 过高或者是组织区域选的不合适)。从这个 50% 的阈值上我们也可以判断 10x 的这个空间转录组技术还是存在一定缺陷的,它允许接近 50% 的 reads 散落在组织以外的区域,说明组织透化这一步想让对应区域的 mRNA 完全都落入对应 spot 点里面去还是很难的。
Mean Reads per Spot:每个 spot 的平均 reads 数,10x 这里采用的是所以测序 reads 总是除以组织上检测到的 spot 数(跟单细胞的统计方法是一样的),理论上来说这样统计是不合理的,因为总的 reads 包括没有比对上的 reads、没有 mapping 到转录本上的 reads、组织区域以外的 spot 上的 reads,所以是不能真实的反应每个 spot 上实际的 reads 数的。
Median Genes per Spot:每个 spot 的基因中位数
Total Genes Detected:检测到的基因总数
Median UMI Counts per Spot:每个 spot 的中位 UMI 数
样本信息区:

Sample ID:样本 id
Chemistry:试剂版本
Slide Serial Number:Slide 信号和区域
Reference Path:参考基因组路径
Transcriptome:基因组转录组版本
Pipeline Version:spaceranger 软件版本
Analysis 区域
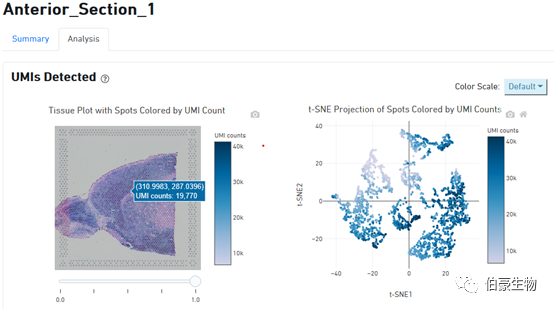
UMI 分布展示:左边是图像上 UMI 的分布,右边是 tsne 降维可视化后的 UMI 的分布,鼠标放置到图像上会现在对应的位置信息和对应 spot 上的 UMI count 数。从这个图我们可以判断 UMI 主要分布在组织的哪些区域,哪些区域没有捕获到 mRNA 或捕获的 mRNA 特别少。
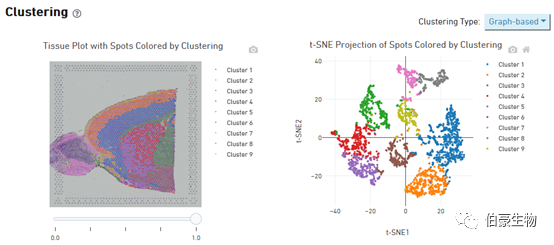
Cluster 的分布展示:左边是 cluster 在组织图像上的分布,右边是 tsne 降维可视化后的 cluster 的分布,鼠标放置到图像上会现在对应的位置信息和对应 spot 上的 cluster 值和该 cluster 占总的 spot 的比例。这个图片上 cluster 分群在组织上的层次关系特别明显。
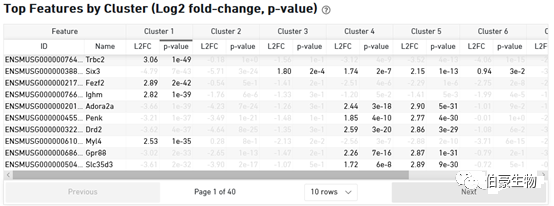
这一部分主要展示亚群的 top 基因的信息,因为不管是单细胞还是空间转录组基本上后面都会自己另外重新分析的,所以这部分和上面的 cluster 分布信息意义不大。
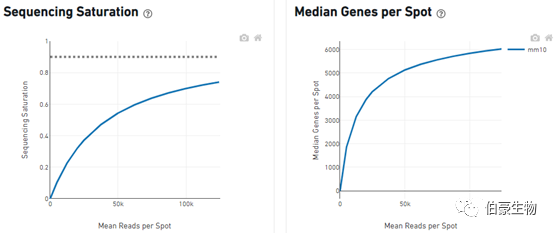
Sequencing Saturation(测序饱和度)
对 reads 进行随机抽样,观察不同 spot 平均 reads 的情况下测序饱和度的分析,一直到实际的测序深度测序饱和度的值,理论上当所有转化的 mRNA 转录本均已测序后,饱和度接近 1.0(100%),虚线表示测序到合理的饱和点位置,也就是说就是测序深度再高也不可能饱和度达到 100%。
Median Genes per Spot(sopt 点的中位基因)
也是对 reads 进行随机抽样,观察不同 spot 平均 reads 的情况下 spot 的中位基因的值,曲线最高点的斜率能反应增加测序深度能得到最大的 spot 的中位基因数。
总结
对于 web_summary 的结果我们大概重点可以从下面几个方面来看数据效果:
1、总的 spot 数,这个其实由组织的大小而定,没有具体好坏的说法;
2、每个 spot 的中位基因数,中位基因数太少说明捕获效果不好,有可能透化步骤条件不够优化,当然也有可能是试剂或芯片的问题;
3、测序饱和度,每个点的 UMI 中位数,sopt 平均 reads 数,饱和度、sopt 平均 reads 数和中位 UMI 数都太低说明测序深度不够,需要加大测序量。
4、基因组的比对率,比对率太低有可能是样品污染;
5、组织 spot 上 reads 的比例,比对太低有可能透化时间不够导致背景 RNA 过高,需要优化透化条件,也有可能组织区域选的不好,这个可以通过 LoupeBrowser 手动选择组织区域。